
Thông tin tờ hướng dẫn sử dụng của thuốc TECENTRIQ
Dưới đây là nội dung tờ hướng dẫn sử dụng của Thuốc TECENTRIQ (Thông tin bao gồm liều dùng, cách dùng, chỉ định, chống chỉ định, thận trọng, dược lý…)
1. Tên hoạt chất và biệt dược:
Hoạt chất : Atezolizumab.
Phân loại: Thuốc chống ung thư.
Nhóm pháp lý: Thuốc kê đơn ETC – (Ethical drugs, prescription drugs, Prescription only medicine)
Mã ATC (Anatomical Therapeutic Chemical): L01XC32.
Brand name: TECENTRIQ
Hãng sản xuất : Roche Diagnostics GmbH. Cơ sở đóng gói và xuất xưởng: F. Hoffmann – La Roche Ltd.
2. Dạng bào chế – Hàm lượng:
Dạng thuốc và hàm lượng
Dung dịch đậm đặc để pha dung dịch tiêm truyền: 60mg/ml.
Thuốc tham khảo:
| TECENTRIQ | ||
| Mỗi ml dung dịch đậm đặc có chứa: | ||
| Atezolizumab | …………………………. | 60 mg |
| Tá dược | …………………………. | vừa đủ (Xem mục 6.1) |

3. Video by Pharmog:
[VIDEO DƯỢC LÝ]
————————————————
► Kịch Bản: PharmogTeam
► Youtube: https://www.youtube.com/c/pharmog
► Facebook: https://www.facebook.com/pharmog/
► Group : Hội những người mê dược lý
► Instagram : https://www.instagram.com/pharmogvn/
► Website: pharmog.com
4. Ứng dụng lâm sàng:
4.1. Chỉ định:
TECENTRIQ được chỉ định để điều trị cho các bệnh nhân mắc ung thư phổi không tế bào nhỏ (NSCLC) di căn mà bệnh vẫn tiến triển trong hoặc sau khi dùng phác đồ hóa trị có chứa platinum.
4.2. Liều dùng – Cách dùng:
Cách dùng :
Không được tiêm tĩnh mạch nhanh.
Không được lắc.
Chuẩn bị
Kiểm tra bằng mắt thường các tiểu phân và sự đổi màu của bao bì và dung dịch pha tiêm truyền trước khi sử dụng. Dung dịch TECENTRIQ không màu hoặc màu vàng nhạt.
Hủy bỏ lọ thuốc nếu dung dịch đục, đổi màu hoặc quan sát thấy các tiểu phân.
Hướng dẫn pha loãng
TECENTRIQ cần được pha chế bởi một cán bộ y tế, sử dụng kĩ thuật vô trùng.
Chuẩn bị dung dịch để truyền như sau:
Lấy 20 mL dung dịch đậm đặc TECENTRIQ từ lọ thuốc
Pha loãng với 250 mL túi polyvinyl clorua (PVC), polyethylene (PE), hoặc polyolefin (PO) có chứa dung dịch natri clorid 0,9% pha tiêm.
Chỉ được pha loãng thuốc với dung dịch tiêm truyền natri clorid 0,9%.
Bảo quản dung dịch sau khi pha loãng
TECENTRIQ không chứa chất bảo quản, do đó, mỗi lọ chỉ được sử dụng một lần.
Liều dùng:
Nguyên tắc chung
TECENTRIQ phải được dùng theo đường truyền tĩnh mạch dưới sự giám sát của một nhân viên y tế có chuyên môn. Không được tiêm tĩnh mạch nhanh.
Cần trao đổi với bác sĩ kê đơn khi có dự định thay thế thuốc này bằng bất kì sinh phẩm nào khác.
Liều khuyến cáo là 1200 mg truyền tĩnh mạch, một lần mỗi 3 tuần. Liều khởi đầu của TECENTRIQ phải được truyền trong 60 phút. Nếu bệnh nhân có khả năng dung nạp tốt với lần truyền đầu tiên, những liều tiếp theo có thể được truyền trong 30 phút.
Thời gian đợt điều trị
Bệnh nhân được điều trị bằng TECENTRIQ cho đến khi thuốc không còn hiệu quả lâm sàng (xem mục Nghiên cứu Hiệu quả/Lâm sàng) hoặc xuất hiện độc tính không thể kiểm soát.
Tạm ngưng hoặc bỏ lỡ một liều
Nếu bị lỡ một liều TECENTRIQ so với dự kiến, cần dùng thuốc ngay khi có thể; không chờ cho đến thời điểm dùng liều kế tiếp. Liệu trình dùng thuốc cần được điều chỉnh để duy trì khoảng cách giữa các liều là 3 tuần.
Điều chỉnh liều
Không khuyến cáo giảm liều TECENTRIQ.
Tạm dừng TECENTRIQ nếu gặp phải bất kỳ trường hợp nào sau đây:
Viêm phổi độ 2 (xem Cảnh báo)
Aspartate aminotransferase (AST) hoặc alanine aminotransferase (ALT) lớn hơn 3 lần và lên đến 5 lần chỉ số trên của giới hạn bình thường (ULN) hoặc bilirubin toàn phần lớn hơn 1,5 lần và lên đến 3 lần so với ULN (xem Cảnh báo)
Tiêu chảy hoặc viêm đại tràng độ 2 hoặc độ 3 (xem Cảnh báo)
Viêm tuyến yên có triệu chứng, suy thượng thận, suy giáp, cường giáp hoặc tăng đường huyết độ 3 hoặc độ 4 (xem Cảnh báo)
Độc tính tiền thị giác độ 2 (xem Cảnh báo)
Viêm tụy độ 2 hoặc độ 3, hoặc tăng nồng độ enzym amylase hoặc lipase độ 3 hoặc độ 4 (lớn hơn hai lần ULN) (xem Cảnh báo)
Nhiễm trùng độ 3 hoặc độ 4 (xem Cảnh báo)
Phản ứng liên quan đến tiêm truyền độ 2 (xem Cảnh báo)
Phát ban độ 3
Viêm cơ tim độ 2 (xem Cảnh báo)
TECENTRIQ có thể được tiếp tục dùng lại ở những bệnh nhân mà tình trạng phản ứng bất lợi được cải thiện đến độ 0-1.
Dừng TECENTRIQ vĩnh viễn nếu gặp phải bất kỳ trường hợp nào sau đây:
Viêm phổi độ 3 hoặc độ 4 (xem Cảnh báo)
Nồng độ AST hay ALT lớn hơn 5 lần ULN hoặc bilirubin toàn phần lớn hơn 3 lần ULN (xem Cảnh báo)
Tiêu chảy hoặc viêm đại tràng độ 4 (xem Cảnh báo)
Viêm tuyến yên độ 4 (xem Cảnh báo)
Hội chứng nhược cơ/bệnh nhược cơ, hội chứng Guillain-Barré hoặc viêm não – màng não (tất cả các mức độ) (xem Cảnh báo)
Độc tính tiền thị giác độ 3 hoặc độ 4 (xem Cảnh báo)
Viêm tụy độ 4 hoặc viêm tụy tái phát bất kỳ độ nào (xem Cảnh báo)
Phản ứng liên quan đến tiêm truyền độ 3 hoặc độ 4 (xem Cảnh báo)
Phát ban độ 4
Viêm cơ tim độ 3 hoặc độ 4 (xem Cảnh báo).
Các hướng dẫn sử dụng liều đặc biệt
Trẻ em
Tính an toàn và hiệu quả của TECENTRIQ trên trẻ em và thiếu niên dưới 18 tuổi chưa được nghiên cứu.
Người cao tuổi
Dựa trên một phân tích dược động học quần thể, không cần điều chỉnh liều TECENTRIQ trên các bệnh nhân ≥65 tuổi (xem mục Sử dụng thuốc trên người cao tuổi và mục Dược động học trên các đối tượng đặc biệt).
Suy thận
Dựa trên một phân tích dược động học quần thể, không cần điều chỉnh liều trên bệnh nhân suy thận (xem mục Dược động học trên các đối tượng đặc biệt).
Suy gan
Dựa trên một phân tích dược động học quần thể, không cần điều chỉnh liều trên bệnh nhân suy gan nhẹ. Chưa có dữ liệu về việc sử dụng thuốc trên bệnh nhân suy gan trung bình hoặc nặng (xem mục Dược động học trên các đối tượng đặc biệt).
4.3. Chống chỉ định:
Chống chỉ định TECENTRIQ trên các bệnh nhân được biết là quá mẫn cảm với TECENTRIQ hoặc bất kì thành phần nào của thuốc.
4.4 Thận trọng:
Thông tin chung
Để cải thiện khả năng truy tìm nguồn gốc của các thuốc sinh phẩm, tên thương mại và số lô của các sản phẩm đã dùng cần được lưu lại rõ ràng (hoặc ghi chú) trong hồ sơ bệnh án.
Viêm phổi liên quan đến miễn dịch
Những trường hợp viêm phổi, có thể dẫn đến tử vong, đã được ghi nhận trong các thử nghiệm lâm sàng với TECENTRIQ (xem Tác dụng ngoại ý, Các thử nghiệm lâm sàng). Bệnh nhân cần được theo dõi các dấu hiệu và triệu chứng của viêm phổi.
Cần ngừng điều trị với TECENTRIQ khi xuất hiện viêm phổi độ 2 và bắt đầu sử dụng prednisone liều 1-2 mg/kg mỗi ngày hoặc tương đương. Nếu các triệu chứng được cải thiện ≤ độ 1, có thể giảm dần liều corticosteroid trong thời gian ≥1 tháng. Có thể điều trị lại với TECENTRIQ nếu viêm phổi được cải thiện ≤ độ 1 trong 12 tuần và liều corticosteroid đã giảm ≤ 10 mg prenisone đường uống mỗi ngày hoặc tương đương. Cần ngừng vĩnh viễn TECENTRIQ khi xuất hiện viêm phổi độ 3 hoặc 4.
Viêm gan liên quan đến miễn dịch
Những trường hợp viêm gan, có thể dẫn đến tử vong, đã được ghi nhận trong các thử nghiệm lâm sàng với TECENTRIQ (xem Tác dụng ngoại ý, Thử nghiệm lâm sàng). Bệnh nhân cần được theo dõi các dấu hiệu và triệu chứng của viêm gan. Cần theo dõi nồng độ aspartat aminotransferase (AST), alanin aminotransferase (ALT) và nồng độ bilirubin trước điều trị và định kì trong quá trình điều trị với TECENTRIQ. Cần xem xét biện pháp xử trí thích hợp đối với các bệnh nhân có bất thường xét nghiệm chức năng gan tại thời điểm trước khi sử dụng thuốc.
Cần ngừng điều trị bằng TECENTRIQ nếu xuất hiện viêm gan độ 2 (ALT hoặc AST >3x chỉ số trên của giới hạn bình thường (ULN) hoặc nồng độ bilirubin máu >1,5x ULN kéo dài nhiều hơn 5-7 ngày và bắt đầu điều trị bằng prednisone liều 1-2 mg/kg mỗi ngày hoặc tương đương. Nếu các xét nghiệm chức gan được cải thiện ≤ độ 1, giảm dần liều corticosteroid trong thời gian ≥1 tháng. Có thể tái sử dụng TECENTRIQ nếu viêm gan được cải thiện ≤ độ 1 trong 12 tuần và liều corticosteroid đã giảm ≤ 10 mg prednisone đường uống mỗi ngày hoặc tương đương. Cần ngừng TECENTRIQ vĩnh viễn khi xuất hiện viêm gan độ 3 hoặc độ 4 (ALT hoặc AST >5,0x ULN hoặc bilirubin máu >3x ULN).
Viêm đại tràng liên quan đến miễn dịch
Những trường hợp tiêu chảy hoặc viêm đại tràng đã được ghi nhận trong các thử nghiệm lâm sàng với TECENTRIQ (xem mục Tác dụng ngoại ý, Các thử nghiệm lâm sàng). Bệnh nhân cần được theo dõi các dấu hiệu và triệu chứng của viêm đại tràng.
Cần ngừng điều trị với TECENTRIQ khi xuất hiện viêm đại tràng (có triệu chứng) hoặc tiêu chảy độ 2 hoặc 3 (tăng số lần đi ngoài ≥4 lần/ngày so với trước điều trị). Đối với trường hợp viêm đại tràng hoặc tiêu chảy độ 2, nếu các triệu chứng kéo dài >5 ngày hoặc tái phát, bắt đầu sử dụng prednisone liều 1-2 mg/kg mỗi ngày hoặc tương đương. Để điều trị viêm đại tràng hoặc tiêu chảy độ 3, cần dùng coricosteroid đường tĩnh mạch (methylprednisolon liều 1-2 mg/kg/ngày hoặc tương đương) và chuyển sang dùng corticosteroid đường uống (prednisone liều 1-2 mg/kg mỗi ngày hoặc tương đương) sau khi các triệu chứng đã cải thiện. Nếu các triệu chứng đã cải thiện ≤ độ 1, cần giảm dần liều corticosteroid trong thời gian ≥1 tháng. Có thể điều trị lại với TECENTRIQ nếu tiêu chảy hoặc viêm đại tràng đã cải thiện ≤ độ 1 trong 12 tuần và liều corticosteroid đã giảm tới ≤ 10 mg prednisone đường uống mỗi ngày hoặc tương đương. Cần ngừng vĩnh viễn TECENTRIQ khi xuất hiện viêm đại tràng hoặc tiêu chảy độ 4 (đe dọa tính mạng; cần can thiệp khẩn cấp).
Bệnh nội tiết liên quan đến miễn dịch
Suy giáp trạng, cường giáp trạng, suy thượng thận, viêm tuyến yên và đái tháo đường tuýp 1, bao gồm các trường hợp nhiễm toan ceton do đái tháo đường, đã được ghi nhận trong các thử nghiệm lâm sàng với TECENTRIQ (xem Tác dụng ngoại ý, Thử nghiệm lâm sàng). Bệnh nhân cần được theo dõi các dấu hiệu và triệu chứng của các bệnh nội tiết. Theo dõi chức năng tuyến giáp trước điều trị và định kì trong suốt quá trình điều trị với TECENTRIQ. Cần xem xét biện pháp xử trí thích hợp đối với các bệnh nhân có xét nghiệm chức năng tuyến giáp bất thường trước khi dùng thuốc.
Các bệnh nhân có xét nghiệm chức năng tuyến giáp bất thường không triệu chứng vẫn có thể dùng TECENTRIQ. Với trường hợp suy giáp có triệu chứng, cần ngừng TECENTRIQ và bắt đầu điều trị thay thế hormon tuyến giáp nếu cần. Suy giáp đơn thuần có thể được điều trị thay thế hormon mà không cần dùng corticosteroid. Với cường giáp có triệu chứng, cần ngừng TECENTRIQ và bắt đầu sử dụng một thuốc kháng giáp trạng như methimazol hoặc carbimazol nếu cần.
Có thể dùng lại TECENTRIQ khi các triệu chứng đã được kiểm soát và chức năng tuyến giáp đã cải thiện.
Với trường hợp suy thượng thận có triệu chứng, cần ngừng sử dụng TECENTRIQ và bắt đầu sử dụng methylprednisolon liều 1-2 mg/kg đường tĩnh mạch mỗi ngày hoặc tương đương. Khi các triệu chứng đã cải thiện, có thể dùng prednisone đường uống với liều 1-2 mg/kg mỗi ngày hoặc tương đương. Nếu các triệu chứng cải thiện ≤ độ 1, có thể giảm dần liều corticosteroid trong ≥1 tháng. Có thể dùng lại TECENTRIQ nếu suy thượng thận được cải thiện ≤ độ 1 trong 12 tuần và liều corticosteroid đã giảm ≤ 10 mg prednisone đường uống mỗi ngày hoặc tương đương, đồng thời bệnh nhân đã được điều trị ổn định bằng phác đồ thay thế hormon (nếu cần).
Cần tạm ngừng điều trị bằng TECENTRIQ trên bệnh nhân viêm tuyến yên độ 2 hoặc độ 3. Cần bắt đầu điều trị với methylprednisolon đường tĩnh mạch liều 1-2 mg/kg mỗi ngày hoặc tương đương và bắt đầu điều trị thay thế hormon nếu cần. Khi các triệu chứng đã được cải thiện ≤ độ 1, giảm dần liều corticosteroid trong thời gian ≥1 tháng. Có thể dùng lại TECENTRIQ nếu triệu chứng cải thiện ≤ độ 1 trong 12 tuần và corticosteroid đã được giảm xuống tương đương ≤ 10 mg prednisone đường uống mỗi ngày hoặc tương đương và bệnh nhân đã được điều trị ổn định bằng phác đồ thay thế (nếu cần). Cần ngưng điều trị bằng TECENTRIQ vĩnh viễn đối với viêm tuyến yên độ 4.
Cần bắt đầu điều trị bằng insulin trên bệnh nhân mắc đái tháo đường tuýp 1. Với trường hợp tăng đường huyết ≥ độ 3 (đường huyết lúc đói >250 mg/dL), cần ngừng sử dụng TECENTRIQ. Có thể dùng lại TECENTRIQ nếu đã kiểm soát được đường huyết với phác đồ thay thế insulin.
Viêm não – màng não liên quan đến miễn dịch
Viêm não – màng não đã được ghi nhận trong các thử nghiệm lâm sàng với TECENTRIQ (xem Tác dụng ngoại ý, Thử nghiệm lâm sàng). Bệnh nhân cần được theo dõi các dấu hiệu và triệu chứng viêm màng não hoặc viêm não.
Cần ngừng vĩnh viễn TECENTRIQ khi xuất hiện viêm màng não hoặc viêm não bất kì độ nào. Cần điều trị với methylprednisolon đường tĩnh mạch với liều 1-2 mg/kg mỗi ngày hoặc tương đương. Chuyển sang dùng prednisone đường uống liều 1-2 mg/kg mỗi ngày hoặc tương đương khi các triệu chứng của bệnh nhân đã cải thiện. Nếu các triệu chứng đã được cải thiện ≤ độ 1, giảm dần liều corticosteroid trong thời gian ≥1 tháng.
Bệnh thần kinh liên quan đến miễn dịch
Hội chứng nhược cơ/bệnh nhược cơ hoặc hội chứng Guillain-Barré, có thể đe dọa tính mạng, đã được ghi nhận trên các bệnh nhân sử dụng TECENTRIQ (xem Tác dụng ngoại ý, Các thử nghiệm lâm sàng). Bệnh nhân cần được theo dõi các triệu chứng của bệnh thần kinh vận động và bệnh thần kinh cảm giác.
Cần ngừng vĩnh viễn TECENTRIQ khi gặp hội chứng nhược cơ/bệnh nhược cơ hoặc hội chứng Guillain-Barré ở bất kì độ nào. Cân nhắc sử dụng corticosteroid toàn thân với prednisone đường uống, liều 1-2 mg/kg mỗi ngày, hoặc tương đương.
Viêm tụy liên quan đến miễn dịch
Viêm tụy, bao gồm tăng nồng độ amylase và lipase huyết thanh, đã được ghi nhận trong các thử nghiệm lâm sàng với TECENTRIQ (xem Tác dụng ngoại ý, Các thử nghiệm lâm sàng). Bệnh nhân cần được theo dõi chặt chẽ các dấu hiệu và triệu chứng gợi ý viêm tụy cấp.
Cần ngừng TECENTRIQ khi nồng độ lipase hoặc amylase tăng ≥ độ 3 (>2,0 chỉ số trên của giới hạn bình thường) hoặc viêm tụy độ 2 hoặc 3 và bắt đầu điều bằng methylprednisolon đường tĩnh mạch với liều 1-2 mg/kg mỗi ngày hoặc tương đương. Khi các triệu chứng đã cải thiện, chuyển sang dùng prednisone đường uống với liều 1-2 mg/kg mỗi ngày hoặc tương đương. Có thể dùng lại TECENTRIQ khi nồng độ lipase hoặc amylase huyết thanh cải thiện ≤ độ 1 trong 12 tháng hoặc các triệu chứng viêm tụy đã thoái lui, đồng thời liều corticosteroid đường uống đã giảm ≤ 10 mg prednisone mỗi ngày hoặc tương đương. Cần ngừng vĩnh viễn TECENTRIQ khi viêm tụy độ 4 hoặc viêm tụy tái phát ở bất kì độ nào.
Viêm cơ tim có liên quan đến miễn dịch
Viêm cơ tim đã được quan sát thấy trong các thử nghiệm lâm sàng với TECENTRIQ (xem Tác dụng ngoại ý, thử nghiệm lâm sàng). Bệnh nhân cần được theo dõi các dấu hiệu và triệu chứng của viêm cơ tim.
Cần tạm ngừng điều trị bằng TECENTRIQ đối với viêm cơ tim độ 2. Cần ngừng vĩnh viễn TECENTRIQ đối với viêm cơ tim độ 3 hoặc 4. Cần xem xét việc bắt đầu điều trị corticosteroid toàn thân.
Phản ứng liên quan đến tiêm truyền
Các phản ứng liên quan đến tiêm truyền thuốc đã được ghi nhận trong các thử nghiệm lâm sàng với TECENTRIQ (xem Tác dụng ngoại ý, Thử nghiệm lâm sàng).
Cần giảm tốc độ truyền hoặc ngừng điều trị trên các bệnh nhân gặp các phản ứng liên quan đến tiêm truyền ở độ 1 hoặc 2. Cần ngừng vĩnh viễn TECENTRIQ trên các bệnh nhân gặp phản ứng tiêm truyền độ 3 hoặc 4. Các bệnh nhân gặp phản ứng liên quan đến tiêm truyền ở độ 1 hoặc 2 có thể tiếp tục dùng TECENTRIQ dưới sự theo dõi chặt chẽ; có thể cân nhắc dự phòng bằng thuốc hạ sốt và thuốc kháng histamin.
Các đối tượng đặc biệt
Bệnh nhân mắc bệnh tự miễn bị loại khỏi các thử nghiệm lâm sàng với TECENTRIQ. Do chưa có đủ dữ liệu, cần thận trọng khi sử dụng TECENTRIQ trên các bệnh nhân mắc bệnh tự miễn sau khi đã đánh giá lợi ích-nguy cơ tiềm tàng.
Độc tính trên phôi thai
Dựa trên cơ chế tác dụng, sử dụng TECENTRIQ có thể gây hại đối với bào thai. Các nghiên cứu trên động vật đã chứng minh rằng ức chế con đường PD-L1/PD-1 có thể dẫn tới tăng nguy cơ đào thải bào thai đang phát triển liên quan đến miễn dịch, dẫn đến chết thai.
Phụ nữ mang thai cần được tư vấn về nguy cơ tiềm tàng đối với thai nhi. Phụ nữ có khả năng mang thai cần được khuyến cáo sử dụng biện pháp tránh thai hữu hiệu trong khi dùng TECENTRIQ và trong 5 tháng kể từ liều cuối cùng (xem mục Phụ nữ và nam giới trong độ tuổi sinh sản và mục Độc tính đối với sự sinh sản).
Nhiễm trùng
Nhiễm trùng nặng, bao gồm nhiễm khuẩn huyết, Herpes não và nhiễm trùng lao dẫn đến xuất huyết sau phúc mạc xảy ra ở những bệnh nhân dùng TECENTRIQ. Theo dõi các dấu hiệu và triệu chứng nhiễm trùng của bệnh nhân và điều trị bằng thuốc kháng sinh đối với các bệnh nhân nghi ngờ hoặc đã bị nhiễm khuẩn. Cần tạm ngừng điều trị bằng TECENTRIQ trên các bệnh nhân nhiễm trùng ≥ độ 3.
Độc tính tiền thị giác
Độc tính tiền thị giác xảy ra ở ≤ 1,0% bệnh nhân điều trị bằng TECENTRIQ.
Lạm dụng và phụ thuộc thuốc
Chưa có dữ liệu được báo cáo.
Sử dụng thuốc trên các đối tượng đặc biệt
Sử dụng thuốc trên trẻ em
Độ an toàn và hiệu quả của TECENTRIQ trên trẻ em và thiếu niên dưới 18 tuổi chưa được nghiên cứu.
Sử dụng thuốc trên người cao tuổi
Không có sự khác biệt toàn bộ về hiệu quả và độ an toàn được ghi nhận giữa các bệnh nhân ≥65 tuổi và những bệnh nhân trẻ hơn (xem mục Hướng dẫn đặc biệt về liều dùng và mục Dược động học trên các đối tượng đặc biệt).
Suy thận
Xem mục Hướng dẫn đặc biệt về liều dùng và mục Dược động học trên các đối tượng đặc biệt.
Suy gan
Xem mục Hướng dẫn đặc biệt về liều dùng và mục Dược động học trên các đối tượng đặc biệt.
Tác động của thuốc trên người lái xe và vận hành máy móc.
Chưa có nghiên cứu về ảnh hưởng của thuốc đến khả năng lái xe và vận hành máy móc.
4.5 Sử dụng cho phụ nữ có thai và cho con bú:
Xếp hạng cảnh báo
AU TGA pregnancy category: D
US FDA pregnancy category: NA
Phụ nữ và nam giới trong độ tuổi sinh sản
Khả năng sinh sản: Dựa trên các nghiên cứu trên động vật, TECENTRIQ có thể làm giảm khả năng sinh sản ở phụ nữ có khả năng sinh sản trong khi đang điều trị (xem phần Sự suy giảm khả năng sinh sản).
Biện pháp tránh thai: Các bệnh nhân nữ có khả năng mang thai cần sử dụng biện pháp tránh thai hữu hiệu trong khi điều trị và tối thiểu 5 tháng sau khi ngừng điều trị với TECENTRIQ (xem mục Thông tin chung và mục Độc tính đối với sự sinh sản).
Thời kỳ mang thai:
Chưa có nghiên cứu lâm sàng về sử dụng TECENTRIQ trên phụ nữ có thai. Không khuyến cáo sử dụng TECENTRIQ trong thai kì trừ khi lợi ích dự kiến đối với người mẹ vượt trội so với nguy cơ tiềm tàng đối với bào thai.
Chuyển dạ và sinh
Sử dụng TECENTRIQ trong thời kì chuyển dạ và sinh con chưa được nghiên cứu.
Thời kỳ cho con bú:
Chưa rõ TECENTRIQ có được bài tiết vào sữa mẹ hay không. Chưa có các nghiên cứu đánh giá tác động của TECENTRIQ đến sự bài tiết sữa hoặc sự có mặt của thuốc này trong sữa. Do chưa xác định được khả năng có hại đối với trẻ nhỏ đang bú mẹ, cần phải quyết định ngừng cho con bú hoặc ngừng điều trị bằng TECENTRIQ.
4.6 Tác dụng không mong muốn (ADR):
Các thử nghiệm lâm sàng
Độ an toàn của Tecentriq đã được thiết lập dựa trên dữ liệu gộp từ 2160 bệnh nhân mắc ung thư biểu mô tiết niệu và ung thư phổi không tế bào nhỏ cùng với dữ liệu bổ trợ trên hơn 8000 bệnh nhân có phơi nhiễm với thuốc trong tất cả các thử nghiệm lâm sàng với nhiều loại khối u. Bảng 4 tóm tắt các phản ứng có hại (ADR) đã được ghi nhận liên quan đến sử dụng Tecentriq.
Các phản ứng có hại được phân loại vào các nhóm với tần suất sau: rất phổ biến (≥1/10), thường gặp (≥1/100 và <1/10), ít gặp (≥1/1000 và <1/100), hiếm gặp (≥1/10000 và <1/1000), rất hiếm gặp (<1/10000).
– xem Bảng 4a & Bảng 4b.


Thông tin bổ sung liên quan đến các phản ứng có hại chọn lọc
Xem Cảnh báo, Thông tin chung liên quan đến quản lý các biến cố sau:
Viêm phổi liên quan đến miễn dịch
Viêm phổi đã xuất hiện trên 3,1% (68/2160) bệnh nhân ung thư biểu mô tiết niệu di căn hoặc ung thư phổi không tế bào nhỏ được điều trị bằng TECENTRIQ. Trong số 68 bệnh nhân này, một trường hợp đã tử vong. Trung vị thời gian khởi phát viêm phổi là 3,5 tháng (dao động từ 3 ngày tới 20,5 tháng). Trung vị khoảng thời gian mắc biến cố này là 1,5 tháng (dao động từ 0 ngày đến 15,1 + tháng; + đại diện cho một giá trị bị khuyết). Viêm phổi dẫn đến phải ngừng thuốc đã xuất hiện trên 10 (0,5%) bệnh nhân. Viêm phổi cần phải dùng corticosteroid xuất hiện trên 1,6% (34/2160) bệnh nhân.
Viêm gan liên quan đến miễn dịch
Viêm gan đã xuất hiện trên 0,3% (7/2160) bệnh nhân mắc ung thư biểu mô tiết niệu di căn hoặc ung thư phổi không tế bào nhỏ được điều trị bằng TECENTRIQ. Trung vị thời gian khởi phát viêm gan là 1,1 tháng (dao động từ 9 ngày đến 7,9 tháng). Trung vị khoảng thời gian mắc biến cố này là 1 tháng (dao động từ 9 ngày đến 1,9+ tháng; + đại diện cho một giá trị bị khuyết). Viêm gan dẫn đến phải ngừng TECENTRIQ đã xuất hiện trên 2 (< 0,1%) bệnh nhân. Viêm gan cần phải dùng corticosteroid đã xuất hiện trên 0,2% (5/2160) bệnh nhân dùng TECENTRIQ.
Viêm đại tràng liên quan đến miễn dịch
Viêm đại tràng đã xuất hiện trên 1,1% (23/2160) bệnh nhân mắc ung thư biểu mô tiết niệu hoặc ung thư phổi không tế bào nhỏ được điều trị bằng TECENTRIQ. Trung vị thời gian khởi phát viêm đại tràng là 4 tháng (dao động từ 15 ngày đến 15,2 tháng). Trung vị khoảng thời gian mắc biến cố này là 1,4 tháng (dao động từ 3 ngày đến 17,8+ tháng; + đại diện cho một giá trị bị khuyết). Viêm đại tràng dẫn đến phải ngừng TECENTRIQ đã xuất hiện trên 5 (0,2%) bệnh nhân. Viêm đại tràng cần phải dùng corticosteroid đã xuất hiện trên 0,5% (10/2160) bệnh nhân dùng TECENTRIQ.
Bệnh nội tiết liên quan đến miễn dịch
Suy giáp trạng đã xuất hiện trên 4,7% (101/2160) bệnh nhân mắc ung thư biểu mô tiết niệu hoặc ung thư phổi không tế bào nhỏ được điều trị bằng TECENTRIQ. Trung vị thời gian khởi phát thiểu năng tuyến giáp là 5,5 tháng (dao động từ 15 ngày đến 31,3 tháng). Cường giáp trạng đã xuất hiện trên 1,7% (36/2160) bệnh nhân mắc ung thư biểu mô tiết niệu hoặc ung thư phổi không tế bào nhỏ được điều trị bằng TECENTRIQ. Trung vị thời gian khởi phát biến cố này là 3,5 tháng (dao động từ 21 ngày đến 31,3 tháng). Suy thượng thận đã xuất hiện trên 0,3% (7/2160) bệnh nhân mắc ung thư biểu mô tiết niệu hoặc ung thư phổi không tế bào nhỏ được điều trị bằng TECENTRIQ. Trung vị thời gian khởi phát suy thượng thận là 5,7 tháng (dao động từ 3 ngày đến 19 tháng). Suy thượng thận cần phải dùng corticosteroid đã xuất hiện trên 0,3% (6/2160) bệnh nhân dùng TECENTRIQ. Viêm tuyến yên đã xuất hiện ở < 0,1% (1/2160) bệnh nhân dùng TECENTRIQ điều trị ung thư biểu mô tiết niệu hoặc ung thư phổi không tế bào nhỏ. Thời gian khởi phát viêm tuyến yên là 13,7 tháng.
Đái tháo đường đã xuất hiện trên 0,3% (6/2160) bệnh nhân mắc ung thư biểu mô tiết niệu hoặc ung thư phổi không tế bào nhỏ được điều trị bằng TECENTRIQ. Thời gian khởi phát suy thượng thận dao động từ 3 ngày đến 6,5 tháng. Đái tháo đường dẫn tới phải ngừng TECENTRIQ đã xuất hiện trên 1 bệnh nhân (< 0,1%).
Viêm não – màng não liên quan đến miễn dịch
Viêm màng não đã xuất hiện trên 0,1% (3/2160) bệnh nhân mắc ung thư biểu mô tiết niệu hoặc ung thư phổi không tế bào nhỏ được điều trị bằng TECENTRIQ. Thời gian khởi phát viêm màng não dao động từ 15 đến 16 ngày. Có 3 bệnh nhân cần phải sử dụng corticosteroid và ngừng TECENTRIQ. Viêm não đã xuất hiện với tỷ lệ < 0,1% (2/2160) bệnh nhân. Thời gian khởi phát viêm não là 14 ngày và 16 ngày. Một trong hai bệnh nhân này cần sử dụng corticosteroid. Viêm não dẫn đến phải ngừng TECENTRIQ đã xuất hiện trên 1 bệnh nhân (< 0,1%).
Bệnh thần kinh liên quan đến miễn dịch
Các bệnh thần kinh, bao gồm hội chứng Guillain-Barré và bệnh mất myelin đa dây thần kinh, đã xuất hiện trên 0,2% (5/2160) bệnh nhân mắc ung thư biểu mô tiết niệu di căn hoặc ung thư phổi không tế bào nhỏ được điều trị bằng TECENTRIQ. Trung vị thời gian khởi phát các bệnh lý này là 7 tháng (dao động từ 18 ngày đến 8,1 tháng). Trung vị khoảng thời gian mắc là 4,6 tháng (dao động từ 0 ngày đến 8,3+ tháng; + đại diện cho một giá trị bị khuyết). Hội chứng Guillain-Barré dẫn đến phải ngừng TECENTRIQ đã xuất hiện trên 1 (< 0,1%) bệnh nhân. Hội chứng Guillain-Barré cần phải điều trị bằng corticosteroid đã xuất hiện với tỷ lệ < 0,1% (2/2160) bệnh nhân.
Viêm tụy liên quan đến miễn dịch
Viêm tụy, bao gồm tăng amylase và lipase, đã xuất hiện trên 0,5% (10/2160) bệnh nhân mắc ung thư biểu mô tiết niệu di căn hoặc ung thư phổi không tế bào nhỏ được điều trị bằng TECENTRIQ. Trung vị thời gian khởi phát viêm tụy là 5,5 tháng (dao động từ 9 ngày đến 16,9 tháng). Trung vị khoảng thời gian mắc biến cố này là 19 ngày (dao động từ 3 ngày đến 11,2+ tháng; + đại diện cho một giá trị bị khuyết). Viêm tụy cần phải điều trị bằng corticosteroid đã xuất hiện với tỷ lệ < 0,1% (2/2160) bệnh nhân dùng TECENTRIQ.
Thông báo cho Bác sĩ những tác dụng không mong muốn gặp phải khi sử dụng thuốc.
4.7 Hướng dẫn cách xử trí ADR:
Các ADR thường hiếm xảy ra và nhẹ, tự hết. Nếu các triệu chứng nặng (lú lẫn, hôn mê ở người suy thận), phải ngừng thuốc ngay Diễn biến thường tốt sau khi ngừng thuốc, ít khi phải thấm phân máu.
4.8 Tương tác với các thuốc khác:
Chưa có nghiên cứu tương tác thuốc-thuốc đặc hiệu với TECENTRIQ.
4.9 Quá liều và xử trí:
Chưa có thông tin về quá liều TECENTRIQ.
5. Cơ chế tác dụng của thuốc :
5.1. Dược lực học:
Nhóm dược lý: Thuốc kháng ung thư, kháng thể đơn dòng globulin miễn dịch G1 (IgG1) có nguồn gốc từ người.
Mã ATC: L01XC323.
Atezolizumab là một kháng thể đơn dòng thuộc nhóm globulin miễn dịch G1 (IgG1) của người được thiết kế dựa trên phần Fc, có khả năng liên kết trực tiếp với PD-L1 và ức chế sự tương tác của phân tử này với các recetor PD-1 và B7.1, dẫn đến giải ức chế sự đáp ứng miễn dịch qua trung gian PD-L1/PD-1, bao gồm tái hoạt hóa đáp ứng miễn dịch chống khối u. Atezolizumab không ảnh hưởng đến tương tác PD-L2/PD-1. Trong các mô hình gây khối u đồng loại trên chuột nhắt trắng, ức chế hoạt tính PD-L1 đã dẫn đến giảm sự phát triển của khối u.
Cơ chế tác dụng:
Liên kết của PD-L1 với các thụ thể PD-1 và B7.1 trên tế bào T dẫn đến ức chế hoạt động của tế bào T gây độc thông qua ức chế sự tăng sinh tế bào T và sản xuất cytokin. PD-L1 có thể biểu hiện trên các tế bào khối u và tế bào miễn dịch thâm nhập vào khối u và có thể góp phần gây ức chế đáp ứng miễn dịch chống khối u trong vi môi trường tại vị trí có khối u.
Atezolizumab là một kháng thể đơn dòng thuộc nhóm globulin miễn dịch G1 (IgG1) của người được thiết kế dựa trên phần Fc, có khả năng liên kết trực tiếp với PD-L1 và ức chế sự tương tác của phân tử này với các recetor PD-1 và B7.1, dẫn đến giải ức chế sự đáp ứng miễn dịch qua trung gian PD-L1/PD-1, bao gồm tái hoạt hóa đáp ứng miễn dịch chống khối u. Atezolizumab không ảnh hưởng đến tương tác PD-L2/PD-1. Trong các mô hình gây khối u đồng loại trên chuột nhắt trắng, ức chế hoạt tính PD-L1 đã dẫn đến giảm sự phát triển của khối u.
[XEM TẠI ĐÂY]
5.2. Dược động học:
Dược động học của TECENTRIQ đã được mô tả trên các bệnh nhân trong nhiều thử nghiệm lâm sàng với các mức liều từ 0,01 mg/kg tới 20 mg/kg mỗi 3 tuần, bao gồm liều cố định 1200 mg. Phơi nhiễm với TECENTRIQ liều tăng tương ứng với khoảng liều từ 1 mg/kg đến 20 mg/kg. Một phân tích quần thể bao gồm 472 bệnh nhân đã mô tả dược động học của TECENTRIQ trong khoảng liều 1-20 mg/kg với mô hình dược động học tuyến tính hai ngăn thải trừ bậc 1. Phân tích dược động học quần thể cho thấy trạng thái ổn định của thuốc đạt được sau 6-9 tuần (2 đến 3 chu kì) dùng liều lặp lại. Tích lũy toàn thân, thể hiện qua các thông số diện tích dưới đường cong (AUC), nồng độ đỉnh (Cmax) và nồng độ đáy (Cmin), lần lượt là 1,91, 1,46 và 2,75 lần.
Dựa trên một phân tích phơi nhiễm, dữ liệu độ an toàn và hiệu quả, các yếu tố sau đã được xác định là không có ảnh hưởng có ý nghĩa lâm sàng đến dược động học của TECENTRIQ: tuổi (21-89), cân nặng, giới tính, trạng thái dương tính với kháng thể kháng trị liệu, nồng độ albumin, tổng khối bướu, chủng tộc và vùng miền, suy thận, suy gan nhẹ, mức biểu hiện PD-L1 hoặc tình trạng ECOG.
Hấp thu
TECENTRIQ được dùng theo đường truyền tĩnh mạch. Sử dụng thuốc theo các đường dùng khác chưa được nghiên cứu.
Phân bố
Một phân tích dược động học quần thể cho thấy thể tích phân bố trong khoang trung tâm (V1) là 3,28 lít và thể tích phân bố đặc trưng tại trạng thái ổn định (Vss) là 6,91 lít.
Chuyển hóa
Chuyển hóa của TECENTRIQ chưa được nghiên cứu trực tiếp. Kháng thể được thải trừ chủ yếu bởi quá trình dị hóa.
Đào thải
Một phân tích dược động học quần thể cho thấy độ thanh thải của TECENTRIQ là 0,200 lít/ngày và thời gian bán thải đặc trưng (t1/2) là 27 ngày.
Dược động học trên các đối tượng đặc biệt
Trẻ em
Chưa có nghiên cứu về dược động học của TECENTRIQ trên trẻ em.
Người cao tuổi
Chưa có nghiên cứu đặc hiệu về dược động học của TECENTRIQ trên người cao tuổi. Ảnh hưởng của tuổi đến dược động học của TECENTRIQ đã được đánh giá trong một phân tích dược động học quần thể. Dựa trên phân tích dữ liệu từ các bệnh nhân có tuổi trong khoảng 21-89 tuổi (n=472) với trung vị tuổi bằng 62, tuổi được xác định không phải là một hiệp biến có ý nghĩa ảnh hưởng đến dược động học của TECENTRIQ. Không ghi nhận sự khác biệt có ý nghĩa lâm sàng về dược động học của TECENTRIQ giữa các bệnh nhân <65 tuổi (n=274), bệnh nhân từ 65 đến 75 tuổi (n=152) và bệnh nhân trên 75 tuổi (n=46) (xem mục Hướng dẫn đặc biệt về liều).
Suy thận
Chưa có các nghiên cứu đặc hiệu được thực hiện với TECENTRIQ trên bệnh nhân suy thận. Trong phân tích dược động học quần thể, không có sự khác biệt có ý nghĩa lâm sàng về độ thanh thải của TECENTRIQ được phát hiện trên các bệnh nhân suy thận nhẹ (mức lọc cầu thận ước tính trong khoảng 60-89mL/phút/1,73m2; n=208) hoặc trung bình (mức lọc cầu thận ước tính trong khoảng 30-59mL/phút/1,73m2; n=116) so với các bệnh nhân có chức năng thận bình thường (mức lọc cầu thận ước tính lớn hơn hoặc bằng 90mL/phút/1,73m2; n=140). Chỉ một vài bệnh nhân có tình trạng suy thận nặng (mức lọc cầu thận ước tính trong khoảng 15-29mL/phút/1,73m2; n=8) (xem mục Hướng dẫn đặc biệt về liều).
Suy gan
Chưa có các nghiên cứu đặc hiệu được thực hiện với TECENTRIQ trên bệnh nhân suy gan. Trong phân tích dược động học quần thể, không có sự khác biệt có ý nghĩa lâm sàng về độ thanh thải của TECENTRIQ giữa các bệnh nhân suy gan nhẹ (bilirubin ≤ chỉ số trên của giới hạn bình thường và AST > chỉ số trên của giới hạn bình thường hoặc bilirubin < 1,0 đến 1,5 lần chỉ số trên của giới hạn bình thường, bất kể giá trị AST, n=71) và các bệnh nhân có chức năng gan bình thường (bilirubin và AST ≤ chỉ số trên của giới hạn bình thường, n=401). Chưa có dữ liệu trên các bệnh nhân suy gan trung bình (bilirubin >1,5 đến 3,0x chỉ số trên của giới hạn bình thường, bất kể giá trị AST) hoặc nặng (bilirubin >3,0x chỉ số trên của giới hạn bình thường, bất kể giá trị AST). Suy gan được định nghĩa theo tiêu chuẩn đánh giá rối loạn chức năng gan của Viện Ung thư Quốc gia (NCI) (xem mục Hướng dẫn đặc biệt về liều).
5.3. Hiệu quả lâm sàng:
Các nghiên cứu về hiệu quả/lâm sàng
Ung thư biểu mô tiết niệu
GO29293
Một thử nghiệm lâm sàng pha II, đa trung tâm, quốc tế, đơn nhóm trị liệu, bao gồm hai đoàn hệ (GO29293) (IMvigor 210) đã được thực hiện trên các bệnh nhân mắc ung thư biểu mô tiết niệu di căn hoặc tiến xa tại chỗ (còn được gọi là ung thư biểu mô tế bào chuyển tiếp bàng quang). Nghiên cứu này đã lựa chọn những bệnh nhân có khối u nguyên phát tại bàng quang, cũng như tại bể thận, niệu quản và niệu đạo. Bệnh nhân bị loại khỏi nghiên cứu nếu có tiền sử mắc bệnh tự miễn, di căn não tiến triển, sử dụng vắc xin sống giảm hoạt lực trong 28 ngày trước thời điểm tuyển chọn, sử dụng các tác nhân kích thích miễn dịch hệ thống trong 6 tuần hoặc các thuốc gây ức chế miễn dịch toàn thân trong 2 tuần trước khi tham gia nghiên cứu. Nghiên cứu này đã lựa chọn được tổng cộng 438 bệnh nhân và có hai đoàn hệ bệnh nhân. Đoàn hệ 1 bao gồm các bệnh nhân ung thư biểu mô tiết niệu di căn hoặc tiến xa tại chỗ chưa được điều trị trước đó và không đủ điều kiện hoặc không phù hợp để điều trị bằng phác đồ hóa trị có cisplatin làm nền tảng hoặc có tình trạng bệnh tiến triển sau 12 tháng điều trị với một phác đồ hóa trị bổ trợ hoặc tân bổ trợ chứa platin. Nhóm đoàn hệ 2 bao gồm các bệnh nhân đã được dùng ít nhất một đợt hóa trị chứa platin để điều trị ung thư biểu mô tiết niệu di căn hoặc tiến xa tại chỗ hoặc các bệnh nhân có bệnh tiến triển trong 12 tháng kể từ khi điều trị bằng phác đồ hóa trị bổ trợ hoặc tân bổ trợ chứa platin. Các mẫu bệnh phẩm khối u được đánh giá tiến cứu sự biểu hiện PD-L1 trên các tế bào miễn dịch thâm nhập vào khối u (IC). Những kết quả này được sử dụng để xác định các phân nhóm biểu hiện PD-L1 cho các phân tích được mô tả dưới đây.
Tecentriq đã được dùng một liều cố định 1200 mg truyền tĩnh mạch vào ngày 1 của mỗi chu kì 21 ngày. Các bệnh nhân thuộc đoàn hệ 1 được điều trị cho đến khi bệnh tiến triển. Các bệnh nhân thuộc đoàn hệ 2 được điều trị cho đến khi không còn lợi ích lâm sàng theo đánh giá của nghiên cứu viên.
Có 119 bệnh nhân được điều trị trong đoàn hệ 1 và 310 bệnh nhân được điều trị trong đoàn hệ 2. Đặc điểm nhân khẩu học và tình trạng ban đầu về khối u của bệnh nhân ở cả hai đoàn hệ có tính đại diện cho các quần thể bệnh nhân trong những bối cảnh điều trị tương ứng. Trung vị tuổi của bệnh nhân trong đoàn hệ 1 là 73, trong đoàn hệ 2 là 66. Đa số các bệnh nhân là nam giới (lần lượt là 81% và 78% trong đoàn hệ 1 và 2) và là người da trắng (91% ở cả hai đoàn hệ).
Trong đoàn hệ 1, sự có mặt của các yếu tố tiên lượng xấu được nhận diện trước khi điều trị là tương tự nhau giữa các phân nhóm bệnh nhân có biểu hiện PD-L1 và nhóm bệnh nhân tham gia nghiên cứu nói chung. Đoàn hệ này bao gồm 24 bệnh nhân (20%) có điểm ECOG bằng 2, 18 bệnh nhân (15%) có hai yếu tố nguy cơ Bajorin (điểm thể trạng ECOG ≥2 và di căn nội tạng), 84 bệnh nhân (71%) bị suy giảm chức năng thận (mức lọc cầu thận < 60mLphút) và 25 bệnh nhân (21%) bị di căn gan.
Trong đoàn hệ 2, 43% bệnh nhân đã dùng ≥2 đợt hóa trị liệu và đã có di căn. 39% bệnh nhân đã được nhận phác đồ hóa trị liệu cuối cùng trong vòng 3 tháng trước khi bắt đầu điều trị bằng Tecentriq. Các phác đồ hóa trị dựa trên platin bệnh nhân đã dùng trước đó bao gồm 73% bệnh nhân được dùng cisplatin, 26% bệnh nhân được dùng carboplatin và không dùng thêm phác đồ dựa trên platin nào khác, 1% bệnh nhân được điều trị bằng phác đồ dựa trên platin khác. Tổng cộng có 78% bệnh nhân có di căn nội tạng. Các yếu tố nguy cơ Bellmunt (điểm ECOG bằng 1, di căn gan tại thời điểm ban đầu và nồng độ hemoglobin < 10g/dL) đã được ghi nhận với tỷ lệ tương ứng là 62%, 31% và 22%.
Tiêu chí hiệu quả chính đối với đoàn hệ 1 là tỷ lệ đáp ứng khách quan (ORR) được đánh giá bởi một nhóm đánh giá độc lập (IRF), sử dụng tiêu chuẩn RECIST v1.1. Tiêu chí hiệu quả chính đối với đoàn hệ 2 là ORR được đánh giá bởi IRF, sử dụng tiêu chuẩn RECIST v1.1, và ORR được đánh giá bởi nghiên cứu viên, sử dụng tiêu chuẩn RECIST cải tiến (mRECIST).
Phân tích chính của đoàn hệ 1 được thực hiện khi tất cả các bệnh nhân đã được theo dõi ít nhất 24 tuần. Trung vị thời gian điều trị của các bệnh nhân thuộc đoàn hệ 1 là 15,0 tuần và trung vị khoảng thời gian theo dõi thời gian sống là 7,6 tháng trên các bệnh nhân có biểu hiện PD-L1 ≥5%, 8,3 tháng trên các bệnh nhân có biểu hiện PD-L1 ≥1% và 8,5 tháng đối với tất cả các bệnh nhân tham gia nghiên cứu. ORR được đánh giá bởi IRF theo tiêu chuẩn RECISTv1.1 đã thể hiện ý nghĩa lâm sàng; tuy nhiên, khi so sánh với tỷ lệ đáp ứng (10%) của một nhóm đối chứng hồi cứu được xác định trước nghiên cứu, ý nghĩa thống kê chưa đạt được với tiêu chí chính. ORR được xác nhận bởi IRF-RECISTv1.1 là 21,9% (CI 95%: 9,3-40,0) trên các bệnh nhân với biểu hiện PD-L1 ≥5%, 18,8% (CI 95%: 10,9-29,0) trên các bệnh nhân với biểu hiện PD-L1 ≥1% và 19,3% (CI 95%: 12,7-27,6) đối với tất cả các bệnh nhân tham gia nghiên cứu. Trung vị khoảng thời gian có đáp ứng (DOR) chưa thể ước lượng được ở các phân nhóm có biểu hiện PD-L1 cũng như tất cả các bệnh nhân tham gia nghiên cứu nói chung. Tiêu chí thời gian sống toàn bộ chưa thể xác định đầy đủ với tỷ lệ biến cố này xấp xỉ 40%. Trung vị thời gian sống toàn bộ đối với tất cả các phân nhóm bệnh nhân (mức biểu hiện PD-L1 ≥5% và ≥1%) và nhóm bệnh nhân tham gia nghiên cứu nói chung là 10,6 tháng.
Một phân tích cập nhật đã được thực hiện với trung vị khoảng thời gian theo dõi thời gian sống của bệnh nhân là 17,2 tháng đối với đoàn hệ 1 và được trình bày tóm tắt trong Bảng 2. Trung vị khoảng thời gian có đáp ứng (DOR) chưa thể ước lượng được ở các phân nhóm có biểu hiện PD-L1 cũng như tất cả các bệnh nhân tham gia nghiên cứu nói chung.
– xem Bảng 1.

Phân tích chính cho đoàn hệ 2 được thực hiện khi tất cả các bệnh nhân đã được theo dõi ít nhất 24 tuần. Trung vị thời gian điều trị của các bệnh nhân thuộc đoàn hệ 2 là 12,3 tuần và trung vị khoảng thời gian theo dõi thời gian sống là 7,6 tháng trên các bệnh nhân có biểu hiện PD-L1 ≥5%, 7,2 tháng trên các bệnh nhân có biểu hiện PD-L1 ≥1% và 7,1 tháng đối với tất cả các bệnh nhân tham gia nghiên cứu. Nghiên cứu này đã đạt được các tiêu chí chính ở tất cả các phân nhóm thuộc đoàn hệ 2, thể hiện ở tỷ lệ đáp ứng khách quan (ORR) được đánh giá bởi IRF theo RECIST v1.1 và bởi nghiên cứu viên theo mRECIST đều khác biệt có ý nghĩa thống kê so với tỷ lệ đáp ứng (10%) của nhóm đối chứng hồi cứu được xác định trước nghiên cứu. ORR được xác nhận bởi IRF-RECISTv1.1 là 27,0% (CI 95%: 18,6-36,8) trên các bệnh nhân với biểu hiện PD-L1 ≥5%, 18,3% (CI 95%: 13,3-24,2) trên các bệnh nhân với biểu hiện PD-L1 ≥1% và 15,1% (CI 95%: 11,3-19,6) đối với tất cả các bệnh nhân tham gia nghiên cứu. ORR được xác nhận bởi nghiên cứu viên theo mRECIST là 26,0% (CI 95%: 17,7-37,7) trên các bệnh nhân với biểu hiện PD-L1 ≥5%, 21,2% (CI 95%: 15,8-27,3) trên các bệnh nhân với biểu hiện PD-L1 ≥1% và 18,3% (CI 95%: 14,2-23,1) đối với tất cả các bệnh nhân tham gia nghiên cứu. Trung vị khoảng thời gian có đáp ứng (DOR) chưa thể ước lượng được ở các phân nhóm có biểu hiện PD-L1 cũng như tất cả các bệnh nhân tham gia nghiên cứu nói chung. Tiêu chí thời gian sống toàn bộ chưa thể xác định đầy đủ với tỷ lệ gặp biến cố này xấp xỉ 45,3%. Chưa ước lượng được trung vị thời gian sống toàn bộ của phân nhóm bệnh nhân có biểu hiện PD-L1 ≥5%, trong khi giá trị này của phân nhóm bệnh nhân có biểu hiện PD-L1 ≥1% và nhóm bệnh nhân tham gia nghiên cứu nói chung lần lượt là 8,0 tháng và 7,9 tháng.
Một phân tích cập nhật đã được thực hiện với trung vị khoảng thời gian theo dõi thời gian sống của bệnh nhân là 21,1 tháng đối với đoàn hệ 2 và được trình bày tóm tắt trong Bảng 3. Trung vị khoảng thời gian có đáp ứng (DOR) chưa thể ước lượng được ở các phân nhóm có biểu hiện PD-L1, trong khi giá trị này ở nhóm các bệnh nhân có biểu hiện PD-L1 <1% là 13,3 tháng (CI 95%: 4,2, NE).
– xem Bảng 2.

PCD4989g
Ngoài ra, hiệu quả của thuốc cũng đã được đánh giá trong một nghiên cứu lâm sàng pha Ia, đa trung tâm, quốc tế, đơn nhóm (PCD4989g). Nghiên cứu được thực hiện trên các bệnh nhân mắc ung thư di căn hoặc tiến xa tại chỗ, bao gồm một đoàn hệ 95 bệnh nhân mắc ung thư biểu mô tiết niệu tại bàng quang được điều trị bằng Tecentriq với trung vị thời gian theo dõi thời gian sống là 29,2 tháng. Trong nghiên cứu này, trung vị của ORR được đánh giá bởi IRF-RECIST v1.1 là 31,8% (CI 95%: 13,9; 54,9) đối với các bệnh nhân có biểu hiện PD-L1 ≥5% và 18,8% (CI 95%: 9,0; 32,6) đối với các bệnh nhân có biểu hiện PD-L1 <5%. Trung vị khoảng thời gian có đáp ứng (DOR) chưa thể ước lượng được ở các bệnh nhân có biểu hiện PD-L1 ≥5%; trị số này ở nhóm các bệnh nhân có biểu hiện PD-L1 <5% là 27,6 tháng (CI 95%: 9,6, NE). Trung vị thời gian sống bệnh không tiến triển theo IRF-RECIST v1.1 ở nhóm bệnh nhân có biểu hiện PD-L1 ≥5% và nhóm bệnh nhân có biểu hiện PD-L1 <5% lần lượt là 2,7 tháng (CI 95%: 1,4; 10,6) và 1,7 tháng (CI 95%: 1,4; 4,0). Trung vị thời gian sống toàn bộ là 9,9 tháng (dao động từ 0,7-35,5+; + đại diện cho một giá trị bị khuyết) đối với nhóm bệnh nhân có biểu hiện PD-L1 <5% và 9,1 tháng đối với nhóm bệnh nhân có biểu hiện PD-L1 ≥5% (dao động từ 0,7 đến 32,8 tháng). Tỷ lệ sống còn toàn bộ tại thời điểm 12 tháng và 24 tháng đối với nhóm tất cả các bệnh nhân tham gia nghiên cứu lần lượt là 46% và 30%.
Ung thư phổi không tế bào nhỏ
GO28915
Một nghiên cứu pha III ngẫu nhiên, nhãn mở, đa trung tâm, quốc tế (GO28915, OAK) đã được thực hiện để đánh giá hiệu quả và độ an toàn của Tecentriq so với doxetaxel trên các bệnh nhân mắc ung thư phổi không tế bào nhỏ di căn hoặc tiến xa tại chỗ có bệnh tiến triển trong hoặc sau một phác đồ hóa trị dựa trên platin. Tổng cộng có 1225 bệnh nhân tham gia nghiên cứu, trong đó nhóm phân tích chính bao gồm 850 bệnh nhân đầu tiên được phân nhóm ngẫu nhiên. Những bệnh nhân đáp ứng tiêu chí lựa chọn được phân tầng dựa trên trạng thái biểu hiện PD-L1 trên các tế bào miễn dịch thâm nhập vào khối u (IC), số đợt hóa trị đã nhận trước đó và đặc điểm mô bệnh học. Bệnh nhân được phân chia ngẫu nhiên (1:1) vào nhóm điều trị bằng Tecentriq hoặc doxetaxel. Nghiên cứu này loại trừ các bệnh nhân có tiền sử mắc bệnh tự miễn, di căn não tiến triển hoặc phụ thuộc corticosteroid, sử dụng vắc xin sống giảm độc lực trong vòng 28 ngày trước nghiên cứu, sử dụng các tác nhân kích thích miễn dịch hệ thống trong 4 tuần hoặc các thuốc ức chế miễn dịch toàn thân trong 2 tuần trước nghiên cứu. Khối u được đánh giá mỗi 6 tuần trong 36 tuần đầu tiên và mỗi 9 tuần sau đó. Mẫu bệnh phẩm khối u được đánh giá tiến cứu để xác định sự biểu hiện PD-L1 trên các tế bào khối u (TC) và IC; kết quả này được sử dụng để xác định các phân nhóm dựa trên biểu hiện PD-L1 trong các phân tích được mô tả dưới đây.
Đặc điểm nhân khẩu học và tình trạng bệnh ban đầu của quần thể bệnh nhân trong phân tích chính là tương tự nhau giữa các nhóm điều trị. Trung vị tuổi của bệnh nhân 64 (dao động từ 33 đến 85), 61% bệnh nhân là nam giới. Đa số các bệnh nhân là người da trắng (70%). Khoảng 3/4 bệnh nhân có ung thư tế bào không vảy (74%), 10% đã được xác định là có đột biến EGFR, 0,2% đã được xác định có tái sắp xếp ALK, 10% có di căn thần kinh trung ương tại thời điểm trước điều trị và đa số bệnh nhân đang hoặc đã từng hút thuốc lá (82%). Điểm thể trạng ECOG là 0 (37%) hoặc 1 (63%). 75% bệnh nhân chỉ nhận một đợt hóa trị có platin làm nền tảng trước đó.
Tecentriq được dùng với liều cố định 1200 mg truyền tĩnh mạch mỗi 3 tuần. Không áp dụng giảm liều trong toàn bộ nghiên cứu. Bệnh nhân được điều trị cho đến khi không còn lợi ích lâm sàng theo đánh giá của nghiên cứu viên. Doxetaxel được dùng với liều 75 mg/m2 truyền tĩnh mạch vào ngày 1 của mỗi chu kì 21 ngày cho đến khi bệnh tiến triển. Xét trên tất cả các bệnh nhân được điều trị trong nghiên cứu, trung vị thời gian điều trị là 2,1 tháng ở nhóm doxetaxel và 3,4 tháng ở nhóm Tecentriq.
Tiêu chí hiệu quả chính là thời gian sống toàn bộ. Những kết quả chính của nghiên cứu này, trong đó trung vị khoảng thời gian theo dõi thời gian sống là 21 tháng, được tóm tắt trong Bảng 3. Đường cong Kaplan-Meier cho thời gian sống toàn bộ ở quần thể bệnh nhân theo chủ ý điều trị (ITT) được trình bày tại Hình 1. Hình 2 tóm tắt kết quả về thời gian sống toàn bộ ở các phân nhóm PD-L1 và theo chủ ý điều trị; kết quả này chứng minh rằng Tecentriq đã thể hiện lợi ích kéo dài thời gian sống toàn bộ ở tất các các phân nhóm bệnh nhân, bao gồm phân nhóm có biểu hiện PD-L1 <1% trên tế bào khối u và tế bào miễn dịch thâm nhập vào khối u.
– xem Bảng 3, Hình 1 & Hình 2.

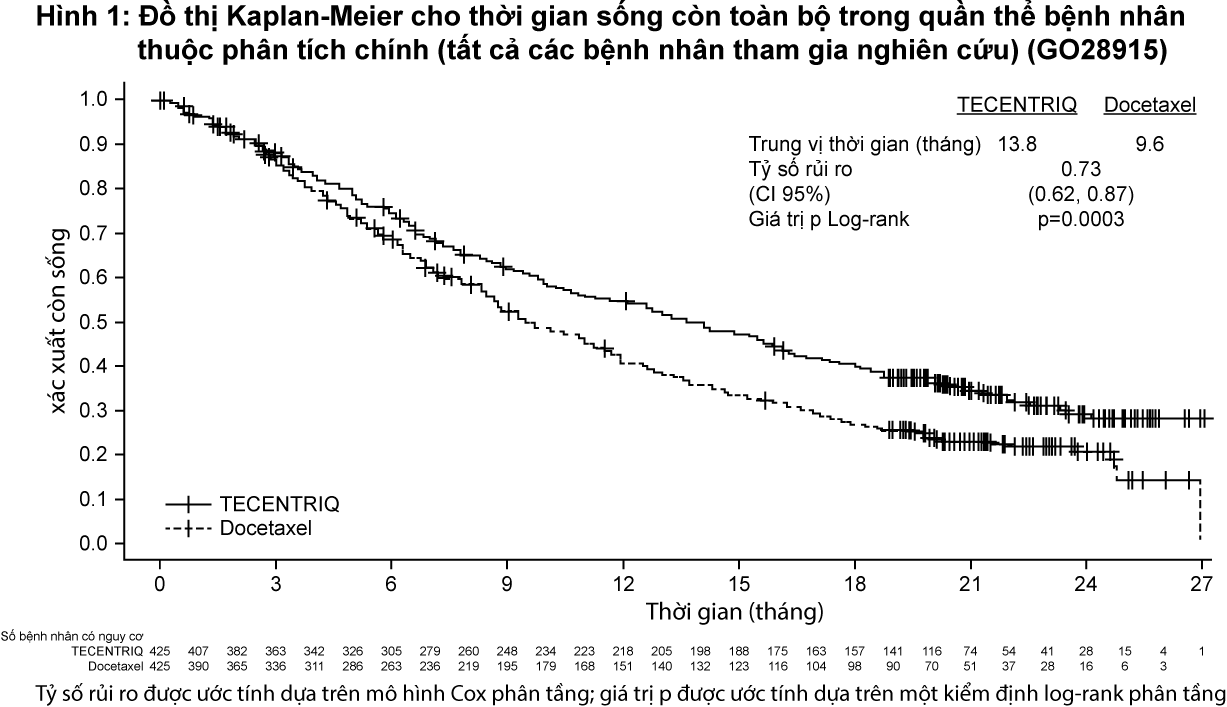
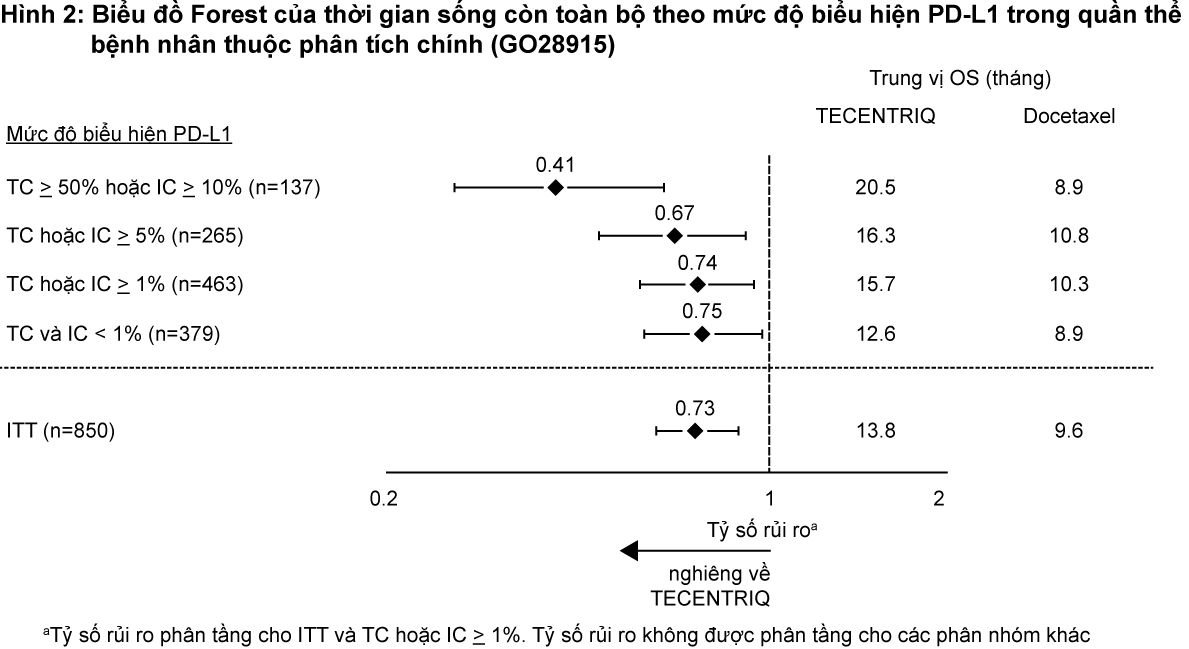
Sự cải thiện thời gian sống còn toàn bộ đã được ghi nhận với Tecentriq so với docetaxel trên cả nhóm bệnh nhân ung thư phổi không tế bào nhỏ không vảy (tỷ số rủi ro [HR]=0,73, CI 95%: 0,60; 0,89; trung vị thời gian sống còn toàn bộ ở nhóm Tecentriq và docetaxel lần lượt là 15,6 tháng và 11,2 tháng) và có vảy (HR=0,73, CI 95%: 0,54; 0,98; trung vị thời gian sống toàn bộ ở nhóm Tecentriq và docetaxel lần lượt là 8,9 tháng và 7,7 tháng). Sự cải thiện thời gian sống còn toàn bộ có tính nhất quán giữa tất cả các phân nhóm bệnh nhân, bao gồm những bệnh nhân có di căn não trước nghiên cứu (HR=0,54, CI 95%: 0,31; 0,94; trung vị thời gian sống còn toàn bộ ở nhóm Tecentriq và docetaxel lần lượt là 20,1 tháng và 11,9 tháng) và những bệnh nhân chưa từng hút thuốc (HR=0,71, CI 95%: 0,47; 1,08; trung vị thời gian sống toàn bộ ở nhóm Tecentriq và docetaxel lần lượt là 16,3 tháng và 12,6 tháng). Tuy nhiên, các bệnh nhân có đột biến EGFR được điều trị bằng Tecentriq không thể hiện sự cải thiện thời gian sống còn toàn bộ khi so sánh với doxetaxel (HR=1,24, CI 95%: 0,71; 2,18; trung vị thời gian sống toàn bộ ở nhóm Tecentriq và docetaxel lần lượt là 10,5 tháng và 16,2 tháng).
Tecentriq kéo dài thời gian đến khi xuất hiện triệu chứng đau ngực được báo cáo bởi bệnh nhân (sử dụng EORTC QLQ-LC13) đã được quan sát với Tecentriq so với doxetacel (HR=0,71, CI 95%: 0,49; 1,05, không có trung vị ở cả hai nhóm). Thời gian đến khi bùng phát các triệu chứng khác của ung thư phổi (bao gồm ho, khó thở và đau cánh tay/vai) (sử dụng EORTC QLQ-LC13) tương tự nhau giữa Tecentriq và doxetacel. Tình trạng sức khỏe tổng thể trung bình và điểm chức năng (bao gồm thể chất, vai trò, xã hội, cảm xúc và nhận thức) được đánh giá bởi EORTC QLQ-C30 không suy giảm có ý nghĩa lâm sàng theo thời gian ở cả hai nhóm điều trị; điều này cho thấy chất lượng cuộc sống và hoạt động chức năng do bệnh nhân báo cáo được duy trì trên các bệnh nhân còn nhận điều trị.
GO28753
Một nghiên cứu pha II, đa trung tâm, quốc tế, được thiết kế ngẫu nhiên, có đối chứng, nhãn mở (GO28753/POPLAR) đã được thực hiện trên các bệnh nhân mắc ung thư phổi không tế bào nhỏ di căn hoặc tiến xa tại chỗ. Tiêu chí hiệu quả chính là thời gian sống còn toàn bộ. Tổng cộng có 287 bệnh nhân được phân chia ngẫu nhiên vào nhóm điều trị bằng Tecentriq hoặc docetaxel. Các nhóm ngẫu nhiên được phân tầng bởi trạng thái biểu hiện PD-L1 trên tế bào miễn dịch thâm nhiễm khối u, số đợt hóa trị liệu đã nhận trước đó và đặc điểm mô bệnh học. Một phân tích cập nhật, trong đó tổng số bệnh nhân đã tử vong là 200 người và trung vị thời gian theo dõi thời gian sống là 22 tháng, cho thấy trung vị thời gian sống toàn bộ là 12,6 tháng trên các bệnh nhân được điều trị bằng Tecentriq và 9,7 tháng trên các bệnh nhân được điều trị bằng docetaxel (HR=0,69, CI 95%: 0,52; 0,92). Tỷ lệ đáp ứng khách quan (ORR) của nhóm Tecentriq và doxetacel là 15,3% so với 14,7% và trung vị thời gian có đáp ứng lần lượt là 18,6 tháng so với 7,2 tháng.
Tính sinh miễn dịch
Tương tự tất cả các protein điều trị khác, cơ thể có khả năng tạo ra đáp ứng miễn dịch với atezolizumab. Trong nghiên cứu GO29293, 49,3% bệnh nhân được xác định dương tính với kháng thể kháng atezolizumab tại một hoặc nhiều thời điểm sau khi dùng thuốc. Trong nghiên cứu GO28915, tỷ lệ phát hiện kháng thể kháng trị liệu (ATA) sau khi dùng thuốc là 30,4%. Nhìn chung, dương tính với kháng thể kháng điều trị có thể ảnh hưởng đến dược động học, hiệu quả hoặc độ an toàn của thuốc.
Các kết quả xét nghiệm tính sinh miễn dịch phụ thuộc nhiều vào một số yếu tố bao gồm độ nhạy và độ đặc hiệu, phương pháp xét nghiệm, cách xử lý mẫu, thời điểm lấy mẫu, các thuốc dùng đồng thời và bệnh lý tiềm ẩn của bệnh nhân. Do đó, so sánh tỷ lệ kháng thể kháng Tecentriq với tỷ lệ kháng thể kháng các chế phẩm khác có thể dẫn đến những kết luận không chính xác.
5.4. Dữ liệu tiền lâm sàng:
Tính sinh ung thư
Các nghiên cứu về khả năng gây ung thư chưa được thực hiện với Tecentriq.
Tính gây đột biến
Các nghiên cứu về khả năng gây đột biến chưa được thực hiện với Tecentriq.
Tính gây giảm khả năng sinh sản
Các nghiên cứu về ảnh hưởng của Tecentriq đến khả năng sinh sản chưa được thực hiện; tuy nhiên, đánh giá cơ quan sinh dục của khỉ đuôi dài đực và cái đã được thực hiện trong nghiên cứu độc tính trường diễn. Tecentriq có ảnh hưởng đến chu kì kinh nguyệt của tất cả khí cái dùng liều 50 mg/kg, đặc trưng bởi một xu hướng chu kì kinh bất thường trong thời gian dùng thuốc và tương ứng với sự thiếu hoàng thể mới trong buồng trứng tại thời điểm quan sát vào cuối nghiên cứu; ảnh hưởng này có khả năng đảo ngược trong giai đoạn nuôi hồi phục (không dùng thuốc). Thuốc không ảnh hưởng đến cơ quan sinh dục nam.
Độc tính đối với sự sinh sản
Chưa có các nghiên cứu về ảnh hưởng của Tecentriq đến khả năng sinh sản hoặc gây dị tật thai nhi trên động vật. Con đường truyền tín hiệu PD-L1/PD-1 đã được xác định là có vai trò thiết yếu đối với sự dung nạp phôi/bào thai và sự sống sót của phôi-bào thai trong thai kì. Sử dụng Tecentriq được dự đoán có thể gây ra các tác dụng bất lợi trong thời kì mang thai và có thể gây nguy hiểm đối với thai nhi trên người, bao gồm chết phôi.
*Lưu ý:
Các thông tin về thuốc trên Pharmog.com chỉ mang tính chất tham khảo – Khi dùng thuốc cần tuyệt đối tuân theo theo hướng dẫn của Bác sĩ
Chúng tôi không chịu trách nhiệm về bất cứ hậu quả nào xảy ra do tự ý dùng thuốc dựa theo các thông tin trên Pharmog.com
6. Phần thông tin kèm theo của thuốc:
6.1. Danh mục tá dược:
Tá dược: Microcrystallin cellulose, copovidon, tinh bột natri glycolat, magnesi stearat, colloidal silica khan vừa đủ 1 viên.
6.2. Tương kỵ :
Tính không tương thích
Không ghi nhận tính không tương thích giữa TECENTRIQ và các túi truyền dịch có bề mặt tiếp xúc bằng polyvinyl chlorid (PVC), polyethylen (PE) hoặc polyolefin. Ngoài ra, không phát hiện tính không tương thích với các màng lọc nội dòng (in-line filter) chứa polyethersulfon hoặc polysulfon và bộ truyền dịch cũng như các dụng cụ phụ trợ truyền dịch khác có chứa PVC, PE, polybutadien hoặc polyetherurethan.
6.3. Bảo quản:
Bảo quản ở nhiệt độ 2°C-8°C.
TECENTRIQ cần được bảo quản tránh ánh sáng.
Không được để đông lạnh. Không được lắc.
Dung dịch pha loãng để truyền cần được dùng ngay. Nếu dung dịch thuốc chưa được dùng ngay, có thể bảo quản ở nhiệt độ 2°C-8°C trong thời gian lên tới 24 giờ hoặc trong 8 giờ ở nhiệt độ phòng (≤ 30°C).
Dùng ngay sau khi chuẩn bị. Nếu dung dịch TECENTRIQ sau khi pha loãng không được sử dụng ngay, có thể bảo quản như sau:
Bảo quản ở nhiệt độ phòng không quá 6 giờ kể từ lúc pha. Thời gian này bao gồm bảo quản ở nhiệt độ phòng dung dịch truyền trong túi truyền và thời gian cho việc tiêm truyền.
Hoặc bảo quản ở nhiệt độ 2°C-8°C trong thời gian không quá 24 giờ. Không để đông lạnh.
6.4. Thông tin khác :
Chưa có thông tin. Đang cập nhật.
6.5 Tài liệu tham khảo:
MIMS Việt Nam
HDSD Thuốc .
7. Người đăng tải /Tác giả:
Bài viết được sưu tầm hoặc viết bởi: Bác sĩ nhi khoa – Đỗ Mỹ Linh.
Kiểm duyệt , hiệu đính và đăng tải: PHARMOG TEAM