1. Tên hoạt chất và biệt dược:
Hoạt chất : Capecitabine
Phân loại: Thuốc chống ung thư, tiền chất của fluorouracil.
Nhóm pháp lý: Thuốc kê đơn ETC – (Ethical drugs, prescription drugs, Prescription only medicine)
Mã ATC (Anatomical Therapeutic Chemical): L01BC06.
Biệt dược gốc: XELODA
Biệt dược: Xelocapec
Hãng sản xuất : Công ty cổ phần dược phẩm Phong Phú – Nhà máy sản xuất dược phẩm Usarichpharm.
2. Dạng bào chế – Hàm lượng:
Dạng thuốc và hàm lượng
Viên nén 500 mg
Thuốc tham khảo:
| XELOCAPEC 500mg | ||
| Mỗi viên nén bao phim có chứa: | ||
| Capecitabine | …………………………. | 500 mg |
| Tá dược | …………………………. | vừa đủ (Xem mục 6.1) |
3. Video by Pharmog:
[VIDEO DƯỢC LÝ]
————————————————
► Kịch Bản: PharmogTeam
► Youtube: https://www.youtube.com/c/pharmog
► Facebook: https://www.facebook.com/pharmog/
► Group : Hội những người mê dược lý
► Instagram : https://www.instagram.com/pharmogvn/
► Website: pharmog.com
4. Ứng dụng lâm sàng:
4.1. Chỉ định:
Ung thư vú: Xelocapec phối hợp với docetaxel được chỉ định để điều trị những bệnh nhân ung thư vú tiến triển tại chỗ hoặc di căn sau khi thất bại với hóa trị liệu độc tế bào. Liệu pháp điều trị trước đây bao gồm anthracycline.
Xelocapec cũng được chỉ định như đơn trị liệu cho điều trị những bệnh nhân ung thư vú tiến triển tại chỗ hoặc di căn sau khi thất bại với chế độ hóa trị bao gồm anthracycline và taxane hoặc cho những ung thư khác mà không có chỉ định dùng anthracycline.
Ung thư đại tràng, ung thư đại trực tràng:
Xelocapec được chỉ định điều trị hỗ trợ cho những bệnh nhân ung thư đại tràng sau phẫu thuật.
Xelocapec được chỉ định điều trị cho những bệnh nhân ung thư đại trực tràng di căn.
Ung thư dạ dày – thực quản: Xelocapec phối hợp với hợp chất platin được chỉ định điều trị bước một cho những bệnh nhân ung thư dạ dày-thực quản tiến triển.
4.2. Liều dùng – Cách dùng:
Cách dùng :
Nên uống Xelocapec với nước trong vòng 30 phút sau khi ăn..
Liều dùng:
Đơn trị:
Ung thư đại trực tràng và ung thư vú
Liều đơn trị được khuyến cáo của Xelocapec là 1250mg/m2 dùng hai lần mỗi ngày (sáng và tối; tương đương với 2500mg/m2 tổng liều mỗi ngày) trong 14 ngày, sau đó là 7 ngày nghỉ thuốc.
Điều trị kết hợp:
Ung thư vú:
Trong trường hợp phối hợp với docetaxel, liều khởi đầu khuyến cáo của Xelocapec là 1250mg/m2 hai lần mỗi ngày trong hai tuần sau đó là một tuần nghỉ, phối hợp với docetaxel 75mg/m2 truyền tĩnh mạch trong vòng 1 giờ mỗi ba tuần.
Theo hướng dẫn sử dụng của docetaxel, thuốc chuẩn bị nên bắt đầu trước khi dùng docetaxel ở những bệnh nhân dùng Xelocapec phối hợp với docetaxel.
Ung thư dạ dày – thực quản và ung thư đại tràng, ung thư đại trực tràng:
Trong trường hợp điều trị phối hợp, liều khuyến cáo của Xelocapec là 800 tới 1000mg/m2 dùng hai lần mỗi ngày trong hai tuần, sau đó 7 ngày nghỉ thuốc, hoặc 625mg/m2 2 lần mỗi ngày khi điều trị liên tục. Các thuốc sinh phẩm khi điều trị kết hợp với Xelocapec không làm ảnh hưởng tới liều khởi đầu của Xelocapec. Ở những bệnh nhân ung thư đại tràng giai đoạn III, điều trị bổ trợ được khuyến cáo trong thời gian tổng cộng 6 tháng.
Thuốc chuẩn bị trước để đảm bảo đủ nước và chống nôn phù hợp theo thông tin sản phẩm cisplatin hoặc oxaliplatin nên dùng trước khi dùng cisplatin hoặc oxaliplatin cho những bệnh nhân điều trị kết hợp Xelocapec với cisplatin hoặc oxaliplatin.
Liều Xelocapec được tính theo diện tích bề mặt cơ thể. Bảng 5 & 6 cho thấy cách tính liều chuẩn và giảm liều cho liều khởi đầu Xelocapec 1250 mg/m2 lẫn 1000 mg/m2.


Điều chỉnh liều trong quá trình điều trị
Điều chỉnh chung:
Độc tính do dùng Xelocapec có thể kiểm soát bằng việc điều trị triệu chứng và/hoặc sự thay đổi liều Xelocapec (ngừng điều trị hoặc giảm liều). Một khi liều đã bị giảm thì không nên tăng ở lần sau đó.
Những độc tính được bác sĩ điều trị cân nhắc là gần như không nghiêm trọng hoặc đe dọa mạng sống thì điều trị có thể được tiếp tục với liều ban đầu mà không cần giảm hoặc ngưng liều.
Không khuyến cáo thay đổi liều cho các tác dụng ngoại ý độ 1. Nên ngừng điều trị với Xelocapec nếu các tác dụng ngoại ý độ 2 hoặc 3 xảy ra. Khi các tác dụng ngoại ý được hồi phục hoặc giảm xuống độ 1, nên bắt đầu điều trị lại với Xelocapec với liều ban đầu hoặc điều chỉnh liều theo bảng 3. Nếu xảy ra các tác dụng ngoại ý độ 4, nên ngừng tạm thời hoặc ngừng vĩnh viễn điều trị cho đến khi các tác dụng ngoại ý được hồi phục hoặc giảm xuống độ 1, và có thể điều trị lại sau đó với liều bằng 50% liều ban đầu. Những bệnh nhân dùng Xelocapec nên được thông báo về việc ngừng điều trị ngay lập tức nếu xuất hiện độc tính mức độ trung bình hoặc xấu hơn. Không được thay thế liều Xelocapec không dùng vì độc tính.
Huyết học:
Không nên điều trị Xelocapec ở những bệnh nhân có lượng bạch cầu đa nhân trung tính ban đầu <1,5 x 109/L và/hoặc tiểu cầu <100 x 109/L. Nếu những đánh giá xét nghiệm đột xuất trong suốt một liệu trình điều trị cho thấy độc tính về huyết học là độ 3 hay độ 4, thì nên được ngừng điều trị Xelocapec.
Bảng 7 cho thấy sự thay đổi liều được khuyến cáo sau khi xuất hiện độc tính với Xelocapec:
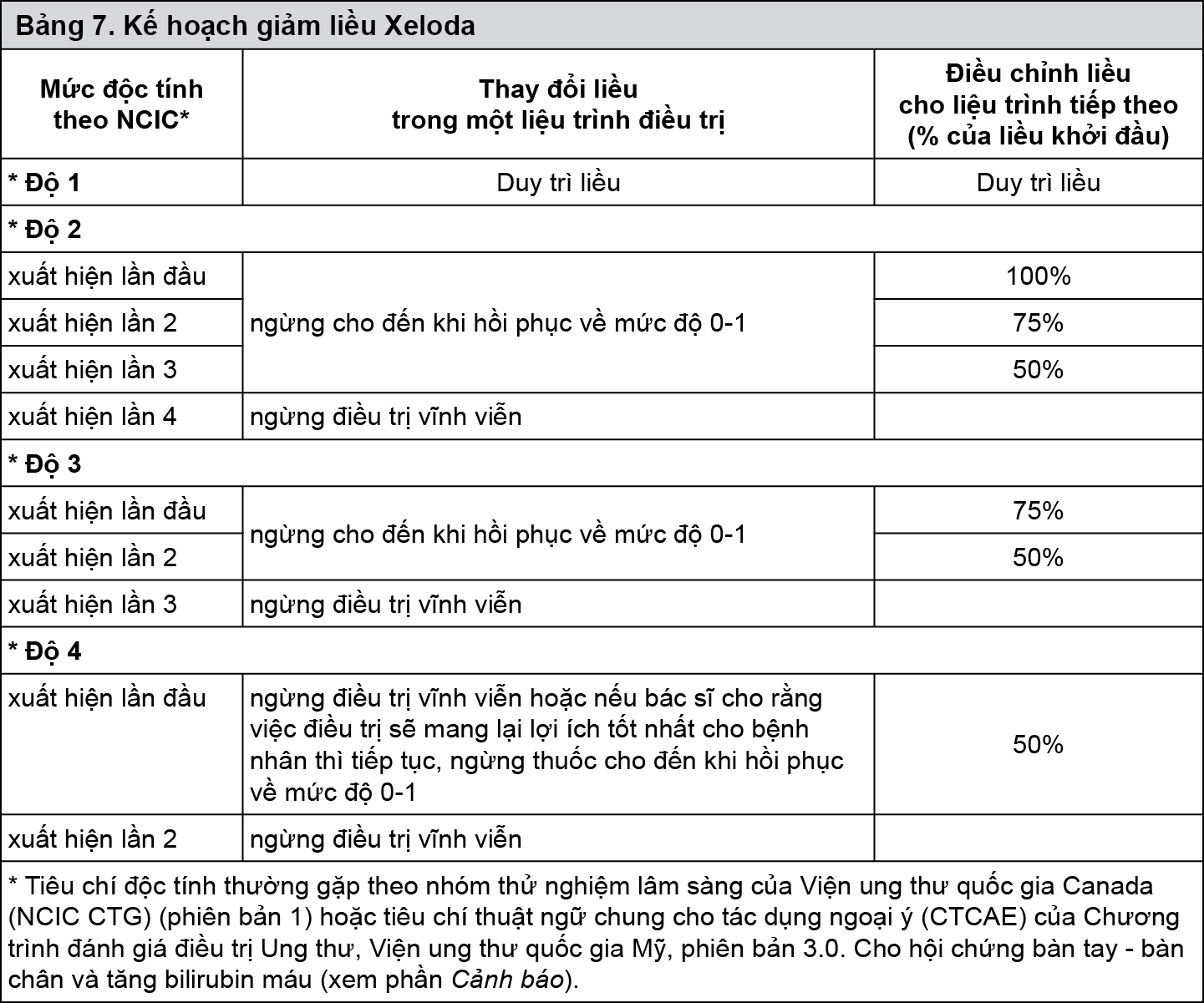
Điều trị kết hợp
Điều chỉnh liều của Xelocapec do độc tính khi Xelocapec kết hợp với các thuốc khác nên dựa theo bảng 7 phía trên cho Xelocapec và theo thông tin kê toa của các thuốc khác một cách thích hợp nhất.
Khi bắt đầu một liệu trình điều trị, nếu hoãn điều trị được chỉ định cho cả Xelocapec hoặc các thuốc khác, thì sau đó nên hoãn tất cả thuốc cho đến khi có những yêu cầu được dùng tất cả thuốc trở lại.
Trong suốt một liệu trình điều trị những độc tính được cân nhắc bởi bác sĩ điều trị mà không liên quan đến Xelocapec (ví dụ: độc tính thần kinh, độc tính ở tai, độc tính thần kinh cảm giác, ứ dịch (tràn dịch màng phổi, tràn dịch màng ngoài tim hoặc cổ chướng, chảy máu, thủng đường tiêu hóa, protein niệu, tăng huyết áp), điều trị Xelocapec nên được tiếp tục và điều chỉnh liều của các thuốc khác dựa vào thông tin kê toa một cách thích hợp.
Nếu các thuốc khác phải ngừng điều trị vĩnh viễn, thì điều trị Xelocapec có thể được bắt đầu lại nếu có các yêu cầu để việc dùng trở lại Xelocapec.
Khuyến cáo này được áp dụng cho tất cả những chỉ định và cho tất cả dân số đặc biệt.
Các hướng dẫn liều dùng đặc biệt
Bệnh nhân bị suy gan do di căn gan
Những bệnh nhân bị suy gan nhẹ tới trung bình do di căn gan, không cần phải điều chỉnh liều khởi đầu. Tuy nhiên, nên theo dõi những bệnh nhân này một cách cẩn thận (xem phần Dược động học trong dân số đặc biệt và phần Cảnh báo). Những bệnh nhân bị suy gan nặng chưa được nghiên cứu.
Bệnh nhân bị suy thận
Capecitabine chống chỉ định ở bệnh nhân suy thận nặng (độ thanh thải creatinine dưới 30mL/phút [Cockroft và Gault] tại thời điểm bắt đầu điều trị). Tỷ lệ phản ứng bất lợi độ 3 hoặc 4 trên bệnh nhân suy thận vừa (độ thanh thải creatinine 30-50mL/phút ở thời điểm bắt đầu điều trị) tăng so với tổng dân số.
Trên những bệnh nhân bị suy thận trung bình (độ thanh thải creatinine 30-50 mL/phút [theo Cockroft và Gault]), tại thời điểm bắt đầu điều trị, người ta khuyến cáo nên giảm còn 75% liều ban đầu nếu liều ban đầu là 1250mg/m2. Trên những bệnh nhân suy thận trung bình, không cần điều chỉnh liều ban đầu nếu liều ban đầu ở mức 1000mg/m2. Trên những bệnh nhân suy thận nhẹ (độ thanh thải creatinine là 51-80 mL/phút ở thời điểm bắt đầu điều trị) không cần điều chỉnh liều ban đầu.
Nên theo dõi cẩn thận và ngừng điều trị ngay lập tức nếu bệnh nhân có các tác dụng ngoại ý độ 2, 3, hoặc 4 trong thời gian điều trị và sau đó điều chỉnh liều như đã chỉ dẫn ở bảng 7 trên (xem phần Dược động học ở dân số đặc biệt). Nên tạm ngừng điều trị Xelocapec, nếu độ thanh thải creatinin giảm dưới 30 mL/phút. Điều chỉnh liều cho những bệnh nhân bị suy thận mức độ trung bình áp dụng cho cả việc điều trị đơn thuần và kết hợp. Để tính toán liều lượng, xem bảng 5 và 6.
Trẻ em: Tính an toàn và hiệu quả của Xelocapec ở trẻ em chưa được thành lập.
Người già:
Không cần điều chỉnh liều khởi đầu khi điều trị Xelocapec đơn trị. Tuy nhiên các tác dụng ngoại ý nghiêm trọng có liên quan đến điều trị độ 3 hoặc 4 xuất hiện thường xuyên hơn ở những bệnh nhân trên 80 tuổi so với những bệnh nhân trẻ hơn.
Khi điều trị Xelocapec kết hợp với các thuốc khác, bệnh nhân lớn tuổi ≥ 65 tuổi từng trải qua những tác dụng thuốc ngoại ý (ADRs) độ 3 và độ 4 nhiều hơn những bệnh nhân trẻ tuổi, ADRs sẽ dẫn đến ngừng tạm thời điều trị. Nên theo dõi cẩn thận những bệnh nhân lớn tuổi.
Khi điều trị Xelocapec kết hợp với docetaxel, tỉ lệ các tác dụng ngoại ý có liên quan đến điều trị độ 3 hoặc 4 và các tác dụng ngoại ý nghiêm trọng tăng được ghi nhận ở những bệnh nhân trên 60 tuổi hoặc hơn. Với những bệnh nhân 60 tuổi hoặc hơn được điều trị phối hợp Xelocapec với docetaxel, nên giảm liều khởi đầu xuống còn 75% (950mg/m2 hai lần mỗi ngày). Để tính toán liều lượng, xem bảng 6.
Với những bệnh nhân 65 tuổi hoặc hơn được điều trị phối hợp Xelocapec với irinotecan, nên giảm liều khởi đầu xuống còn 800mg/m2 hai lần mỗi ngày.
4.3. Chống chỉ định:
Xelocapec được chống chỉ định trên những bệnh nhân được biết là quá mẫn với capecitabine hoặc với bất kỳ thành phần nào của thuốc.
Xelocapec được chống chỉ định trên những bệnh nhân có tiền sử các phản ứng nghiêm trọng và không dự đoán trước với fluoropyrimidine hoặc được biết là quá mẫn với fluorouracil.
Phụ nữ có thai và cho con bú.
Bệnh nhân giảm bạch cầu, bạch cầu trung tính và tiểu cầu nặng.
Không nên dùng Xelocapec cùng với sorivudine hoặc các chất tương tự có liên quan về mặt hóa học, như là brivudine (xem phần Tương tác).
Xelocapec được chống chỉ định trên những bệnh nhân bị suy gan nặng (Child Pugh C).
Xelocapec được chống chỉ định trên những bệnh nhân bị suy thận nặng (độ thanh thải creatinine giảm dưới 30 mL/phút).
Nếu có chống chỉ định của bất kỳ thuốc nào trong điều trị kết hợp, thì không dùng thuốc đó.
4.4 Thận trọng:
Lưu ý đặc biệt
Tiêu chảy: Xelocapec có thể gây tiêu chảy, đôi khi nặng. Bệnh nhân bị tiêu chảy nặng nên được theo dõi cẩn thận và nếu bị mất nước, nên bù nước và điện giải. Nên bắt đầu điều trị tiêu chảy chuẩn (ví dụ loperamide), bằng các thuốc thích hợp càng sớm càng tốt. Giảm liều nên được áp dụng khi cần thiết.
Mất nước: Mất nước nên được ngăn chặn hoặc điều chỉnh đúng lúc. Những bệnh nhân chán ăn, suy nhược, buồn nôn, nôn hoặc tiêu chảy có thể mất nước một cách nhanh chóng.
Nếu mất nước độ 2 (hoặc cao hơn) xuất hiện, nên ngừng điều trị Xelocapec ngay lập tức và mất nước được hiệu chỉnh. Không nên điều trị lại cho đến khi bệnh nhân vẫn còn mất nước và bất kỳ nguyên nhân sớm nào gây ra phải được hiệu chỉnh hoặc kiểm soát. Nên điều chỉnh liều cho những tác dụng ngoại ý đến sớm khi cần thiết.
Thận trọng
Độc tính trên tim mạch:
Ghi nhận được ở Xelocapec cũng tương tự độc tính ghi nhận được ở các fluorinated pyrimidine khác. Những độc tính này bao gồm nhồi máu cơ tim, đau thắt ngực, rối loạn nhịp tim, ngừng tim, suy tim và thay đổi về điện tâm đồ. Những tác dụng ngoại ý này thường gặp hơn ở những bệnh nhân có tiền sử bệnh mạch vành.
Hiếm, không mong đợi, các tác dụng ngoạiý nghiêm trọng: ( ví dụ như Viêm miệng, tiêu chảy, giảm bạch cầu trung tính và độc tính về thần kinh) kết hợp với sự thiêu hụt hoạt tính của dihydropyrimidine dehydrogenase (DPD) duge cho la do 5— FU. Do vậy không thê loại trừ có sự liên kết giữa việc giảm hàm lượng DPD và tăng tác động độc của 5 — FU gay tác hại một cách tiêm ân.
Xelocapec có thể gây ra hội chứng bàn tay-bàn chân (rối loạn cảm giác đỏ da lòng bàn tay-bàn chân hoặc gây ban đỏ đầu chi do hóa trị liệu) là độc tính da. Hội chứng bàn tay, bàn chân dai dẳng hoặc nghiêm trọng (từ mức độ 2 trở lên) có thể dẫn đến mất vân tay, điều này làm ảnh hưởng đến việc nhận diện bệnh nhân. Với những bệnh nhân bị di căn đang điều trị Xelocapec đơn thuần, trung vị thời gian để xuất hiện độc tính là 79 ngày (trong khoảng 11 đến 360 ngày), mức độ từ độ 1 đến 3.
Hội chứng bàn tay-bàn chân độ 1 được xác định bởi tê, rối loạn cảm giác/dị cảm, kiến bò, hoặc ban đỏ ở tay và/hoặc chân và/hoặc không cảm thấy thoải mái nhưng không ảnh hưởng đến các hoạt động bình thường.
Độ 2 được xác định là ban đỏ và sưng tay và/hoặc chân gây đau và/hoặc không thoải mái ảnh hưởng đến các hoạt động của cuộc sống hàng ngày của bệnh nhân.
Độ 3 được xác định là sự tróc vảy da, loét, mụn nước hoặc đau nhiều ở tay và/hoặc chân và/hoặc rất khó chịu khiến bệnh nhân không thể làm việc hoặc thực hiện các hoạt động thông thường hàng ngày.
Nếu xảy ra hội chứng bàn tay-bàn chân độ 2 hoặc 3, nên ngừng dùng Xelocapec cho đến khi các trường hợp hồi phục hoặc giảm xuống độ 1. Sau khi xuất hiện hội chứng bàn tay-bàn chân độ 3, nên giảm liều tiếp theo của Xelocapec (xem phần Liều lượng và Cách dùng). Khi sử dụng kết hợp Xelocapec và cisplatin, sử dụng vitamin B6 (pyridoxine) không được khuyến khích cho triệu chứng hoặc điều trị dự phòng thứ phát của hội chứng bàn tay-bàn chân, bởi vì các báo cáo đã công bố có thể làm giảm hiệu quả của cisplatin.
Capecitabine có thể gây tăng bilirubin máu. Nên ngừng dùng Xelocapec nếu bilirubin tăng >3,0 x ULN (giới hạn trên của mức bình thường) có liên quan đến điều trị hoặc men gan aminotransferase (ALT, AST) tăng >2,5 x ULN có liên quan đến điều trị. Có thể bắt đầu điều trị lại khi bilirubin giảm xuống ≤3,0 x ULN hoặc aminotransferase gan giảm xuống ≤2,5 x ULN.
Trong một nghiên cứu tương tác thuốc với warfarin liều đơn, AUC trung bình của S-warfarin tăng nhiều (+ 57%). Những kết quả này cho thấy sự tương tác thuốc có thể do capecitabine ức chế hệ thống isoenzyme cytochrome P450 2C9. Những bệnh nhân dùng Xelocapec kết hợp với thuốc chống đông dạng uống dẫn xuất của coumarin phải được theo dõi cẩn thận về hiệu quả chống đông của thuốc (INR hoặc thời gian prothrombin) và điều chỉnh liều thuốc chống đông cho phù hợp.
Cảnh báo chung: Bệnh nhân được điều trị bằng Xelocapec nên được theo dõi cẩn thận về độc tính. Phần lớn các tác dụng ngoại ý có thể hồi phục và không yêu cầu phải ngừng thuốc vĩnh viễn, mặc dù vẫn cần phải ngừng thuốc hoặc giảm liều (xem phần Liều lượng và Cách dùng).
Tác động của thuốc trên người lái xe và vận hành máy móc.
Capecitabin có thể gây chóng mặt, mệt mỏi và buồn nôn; bệnh nhân dùng thuốc này cần thận trọng khi lái xe hoặc vận hành máy.
4.5 Sử dụng cho phụ nữ có thai và cho con bú:
Xếp hạng cảnh báo
AU TGA pregnancy category: D
US FDA pregnancy category: D
Thời kỳ mang thai:
Ảnh hưởng tới phụ nữ có thai mức độ D.
Không có nghiên cứu nào trên phụ nữ có thai dùng Xelocapec; tuy nhiên, dựa vào các đặc tính dược học và độc tính, có thể thấy rằng Xelocapec có thể gây hại cho thai nếu được dùng cho phụ nữ có thai. Trong các nghiên cứu độc tính sinh sản trên động vật, dùng capecitabine gây chết phôi và quái thai. Những bằng chứng này có thể xảy ra ở những dẫn xuất của fluoropyrimidine. Capecitabine được cho rằng có khả năng gây quái thai ở người. Capecitabine chống chỉ định trong thời kỳ mang thai. Không nên dùng Xelocapec trong khi mang thai. Nếu dùng Xelocapec trong khi mang thai hoặc nếu bệnh nhân có thai trong khi dùng thuốc này, phải thông báo cho bệnh nhân biết nguy cơ tiềm ẩn cho thai. Nên khuyên những phụ nữ trong độ tuổi sinh đẻ tránh mang thai trong khi điều trị với Xelocapec.
Thời kỳ cho con bú:
Không biết liệu thuốc được bài tiết qua sữa mẹ hay không. Trong một nghiên cứu cho chuột đang cho con bú uống Xelocapec liều đơn, một lượng đáng kể các chất chuyển hóa của capecitabine được bài tiết trong sữa. Nên ngừng cho con bú trong quá trình điều trị Xelocapec.
4.6 Tác dụng không mong muốn (ADR):
Hệ Tiêu hóa: khô miệng, đầy bụng, tiêu chảy, các tác dụng ngoại ý có liên quan đến viêm/loét niêm mạc như viêm thực quản, dạ dày, viêm tá tràng, viêm đại tràng, chảy máu dạ dày.
Tim mạch: phù chỉ dưới, đau ngực do tim bao gồm đau thắt ngực, bệnh cơ tim, thiếu máu cục bộ/ nhồi máu co tim, suy tim, đột tử, nhịp tim nhanh, loạn nhĩ bao gồm rung nhĩ và ngoại tâm thu thất.
Thần Kinh: rôi loạn vị giác, mắt ngủ, nhằm lẫn, bệnh não, và các dấu hiệu về tiểu não như thất điều tiêu não, loạn vận ngôn, giảm thăngbằng, phối hợp bất thường.
Nhiễm trùng và nhiễm ký sinh trùng: các phản ứng phụ liên quan tới suy tủy, tổn thương hệ miễn dịch, và/hoặc thủng niêm mạc, như các nhiễm trùng tại chỗ và nhiễm trùng toàn thân có thể gây tử vong ( bao gôm các nguyên nhân do vi khuẩn, vi rút, nắm) và nhiễm khuẩn.
Máu và bạch huyết: thiếu máu, suy tủy ( được ghỉ nhận là một tác dụng ngoại ý), giảm ba dòng huyết cầu.
Da và tổ chức dưới da: Nút da, ngứa, tróc da khu trú, các rối loạn móng, các phản ứng nhạy cảm với ánh sáng, hội chứng xuất hiện lại các tác dụng phụ giống như khi xạ trị, bong móng, móng dễ gãy, loạn dưỡng móng.
Toàn thân và tình trạng bản thân: suy nhược, đau chân, ngủ lịm, đau ngực (không do tim).
Mắt: viêm kết mạc, kích thích mắt.
Hô hấp: khó thở, ho.
Cơ xương: đau lưng, đau cơ, đau khớp.
Các rối loạn tâm thân: trầm cảm.
Suy gan và viêm gan tt mật được ghi nhận trong các thử nghiệm lâm sàng và sau khi lưu hành trên thị trường. Mối quan hệ nhân quả với việc điều trị bằng Capecitabin chưa được thiết lập.
Thông báo cho Bác sĩ những tác dụng không mong muốn gặp phải khi sử dụng thuốc.
4.7 Hướng dẫn cách xử trí ADR:
Đa số các ADR của capecitabin là hồi phục được và không cần phải ngừng thuốc. Nếu bị nặng thì phải giảm liều (xem bảng hướng dẫn điều chỉnh liều theo mức độ bị độc).
Hay gặp rối loạn tiêu hóa khi dùng capecitabin, nhất là ở người cao tuổi. Có thể dùng các thuốc chống ỉa chảy thông thường như loperamid. Phải bồi phụ nước và điện giải nếu bị ỉa chảy nặng, mất nước. Dùng thuốc chống nôn nếu bị nôn.
Phải kiểm tra huyết học trước khi dùng capecitabin. Nếu thấy bạch cầu trung tính giảm (< 1,5 x 109/lít) và/hoặc tiểu cầu giảm (< 100 x 109/lít) thì không được dùng capecitabin. Trong khi điều trị, nếu thấy bạch cầu trung tính giảm (< 1,0 x 109/lít) và/hoặc tiểu cầu giảm (< 75 x 109/lít) thì phải ngừng thuốc. Nếu có suy tủy: Có thể dùng các yếu tố kích thích tạo máu như filgrastim, sargramostim, truyền máu, truyền tiểu cầu… Bệnh nhân bị giảm bạch cầu trung tính nặng phải được cách ly để tránh nhiễm khuấn. Có thể phải truyền tủy cho người bị suy tủy nặng. Phải phòng giảm bạch cầu trung tính ở người có nguy cơ cao.
Sốt có giảm bạch cầu trung tính: Phải cấy máu và dùng kháng sinh đường tĩnh mạch với người có nguy cơ cao, dùng theo đường uống với người có nguy cơ thấp.
Giám sát chặt chẽ chức năng gan, nhất là ở người bị di căn ở gan vì có nguy cơ tăng bilirubin huyết nghiêm trọng.
Phải thường xuyên theo dõi điện tâm đồ, nhất là ở người có tiền sử bệnh mạch vành để phòng nhồi máu cơ tim, thiếu máu cục bộ cơ tim, cơn đau thắt ngực, loạn nhịp, ngừng tim, đột tử.
Viêm miệng: Rửa, xúc miệng bằng nước muối, bicarbonat. Nếu bị đau thì có thể dùng thuốc giảm đau tại chỗ; nếu đau nặng thì dùng thuốc giảm đau toàn thân. Chú ý dùng các thuốc có tác dụng tại chỗ để phòng nhiễm nấm và nhiễm khuấn miệng.
Phải đi khám mắt nếu thấy thị lực giảm hoặc có các triệu chứng nặng ở mắt trong quá trình dùng capecitabin.
Điều trị các biểu hiện ngộ độc khác tùy theo biểu hiện và mức độ biểu hiện.
4.8 Tương tác với các thuốc khác:
Thuốc chống đông máu coumarin: Đã ghi nhận các thông số đông máu thay đổi và/hoặc chảy máu ở những bệnh nhân dùng Xelocapec cùng với các thuốc chống đông dẫn xuất coumarin như warfarin và phenprocoumon. Những tác dụng phụ này xảy ra trong vài ngày và kéo dài tới vài tháng sau khi điều trị Xelocapec và trong một số ít trường hợp, trong một tháng sau khi ngừng Xelocapec. Trong một nghiên cứu tương tác lâm sàng, sau khi dùng warfarin liều đơn 20mg, việc điều trị Xelocapec làm tăng AUC của S-warfarin khoảng 57% và giá trị INR tăng 91%. Những bệnh nhân dùng các thuốc chống đông dẫn xuất của coumarin cùng với Xelocapec nên được theo dõi cẩn thận về những thay đổi các thông số đông máu của họ (PT hoặc INR) và phải điều chỉnh liều thuốc chống đông cho phù hợp.
Cơ chất của Cytochrome P-450 2C9: Không có các nghiên cứu tương tác thuốc với thuốc chính thức được tiến hành với capecitabine và các thuốc khác được biết là chuyển hóa bởi isoenzyme cytochrome P450 2C9. Nên theo dõi cẩn thận khi Xelocapec dùng cùng với những thuốc này.
Phenytoin: Nồng độ huyết tương của phenytoin tăng được ghi nhận trong khi dùng Xelocapec cùng với phenytoin. Chưa có nghiên cứu tương tác thuốc với thuốc chính thức nào được tiến hành với phenytoin, nhưng cơ chế tương tác thuốc được cho là capecitabine ức chế hệ thống isoenzyme CYP2C9 (xem phần các thuốc chống đông coumarin). Những bệnh nhân dùng phenytoin cùng với Xelocapec nên được theo dõi cẩn thận vì nồng độ huyết tương của phenytoin tăng.
Tương tác thuốc-thức ăn: Trong tất cả các thử nghiệm lâm sàng, bệnh nhân được hướng dẫn uống Xelocapec trong vòng 30 phút sau khi ăn. Vì dữ liệu hiện nay về tính an toàn và hiệu quả dựa trên việc dùng cùng với thức ăn, nên sử dụng Xelocapec cùng với thức ăn.
Thuốc kháng acid: Tác động của hydroxide nhôm và thuốc kháng acid có chứa magnesium hydroxide lên dược động học của capecitabine đã được nghiên cứu trên những bệnh nhân ung thư. Nồng độ huyết tương của capecitabine và một chất chuyển hóa (5’DFCR) tăng ít; không thấy tác động lên ba chất chuyển hóa chính (5’DFUR, 5-FU và FBAL).
Acid folinic/acid folic: Một nghiên cứu phối hợp capecitabine và acid folinic đã chỉ ra rằng acid folinic không ảnh hưởng nghiêm trọng lên dược động học của capecitabine và các chất chuyển hóa của nó. Tuy nhiên, acid folinic có tác động lên dược lực học của capecitabine và độc tính của capecitabine có thể tăng lên bởi acid folic: liều dung nạp tối đa (MTD) của capecitabine điều trị đơn độc khi sử dụng phác đồ không liên tục là 3000mg/m2 mỗi ngày, trong khi đó liều dung nạp tối đa của capecitapine chỉ là 2000mg/m2 mỗi ngày khi sử dụng kết hợp với acid folinic (30mg uống). Độc tính tăng cường có thể liên quan khi chuyển đổi từ phác đồ 5-FU/LV sang phác đồ capecitabine. Điều này cũng có thể liên quan với việc bổ sung acid folic do thiếu hụt folate do sự giống nhau giữa acid folinic và acid folic.
Sorivudine và các thuốc tương tự: Tương tác thuốc-thuốc có ý nghĩa lâm sàng giữa sorivudine và 5-FU, do sorivudine ức chế dihydropyrimidine dehydrogenase, đã được mô tả trong y văn. Tương tác này, dẫn tới tăng độc tính của dihydropyrimidine dehydrogenase, có thể nguy hiểm đến tính mạng. VÌ vậy, không nên dùng Xelocapec với sorivudine hoặc các thuốc tương tự có liên quan về mặt hóa học, như brivudine (xem phần Chống chỉ định). Cần đợi ít nhất 4 tuần sau khi kết thúc điều trị với sorivudine hoặc các thuốc tương tự có liên quan về mặt hoá học như brivudine trước khi bắt đầu trị liệu với Xelocapec.
Oxaliplatin: Không có khác biệt có ý nghĩa lâm sàng khi tiếp xúc với capecitabine hoặc các chất chuyển hóa, platinum tự do hoặc platinum toàn phần xuất hiện khi capecitabine và oxaliplatin được dùng trong điều trị kết hợp, có hoặc không có bevacizumab.
Bevacizumab: Không có tác động có ý nghĩa lâm sàng của bevacizumab lên các thông số dược động học của capecitabine hoặc các chất chuyển hóa của nó.
4.9 Quá liều và xử trí:
Biểu hiện của quá liều cấp tính bao gồm buồn nôn, nôn, tiêu chảy, viêm niêm mạc, kích thích và chảy máu đường tiêu hóa, và suy tủy.
Việc điều trị quá liều nên bao gồm điều trị thông thường và các can thiệp về y khoa hỗ trợ nhằm chữa trị những triệu chứng lâm sàng đang hiện diện và phòng những biến chứng có thể xảy ra.
5. Cơ chế tác dụng của thuốc :
5.1. Dược lực học:
Capecitabine là dẫn xuất fluoropyrimidine carbamate được điều chế để dùng đường uống, là thuốc độc tế bào được hoạt hóa bởi khối u và chọn lọc trên khối u.
Capecitabine không phải là chất độc tế bào trên in vitro. Tuy nhiên, trên in vivo, thuốc được biến đổi liên tiếp thành chất gốc độc tế bào là 5-fluorouracil (5-FU), chất này sẽ được chuyển hoá tiếp.
Sự hình thành 5-FU tại khối u nhờ xúc tác một cách tối ưu của yếu tố tạo mạch liên quan tới khối u là thymidine phosphorylase (dThdPase), nhờ đó làm giảm tối đa mức độ của mô lành với 5-FU trong cơ thể.
Sự biến đổi sinh học tuần tự của men từ capecitabine thành 5-FU dẫn tới nồng độ của 5-FU cao hơn trong mô khối u. Sau khi cho bệnh nhân bị ung thư đại trực tràng (N=8) uống capecitabine, tỉ số nồng độ của 5FU ở khối u đại trực tràng so với các mô gần kề là 3,2 (dao động từ 0,9 đến 8,0). Tỉ số nồng độ ở khối u so với huyết tương là 21,4 (dao động từ 3,9 đến 59,9) trong khi tỉ số ở các mô khỏe mạnh so với huyết tương là 8,9 (dao động từ 3,0 đến 25,8). Hoạt tính của thymidine phosphorylase cao hơn gấp 4 lần ở khối u đại trực tràng so với mô bình thường bên cạnh.
Một vài khối u ở người, như ung thư vú, dạ dày, đại trực tràng, cổ tử cung, và buồng trứng, có nồng độ thymidine phosphorylase cao hơn (có khả năng chuyển 5′-DFUR [5′-deoxy-5-fluorouridine] thành 5-FU) so với các mô bình thường tương ứng.
Các tế bào bình thường và các tế bào khối u chuyển hóa 5-FU thành 5-fluoro-2-deoxyuridine monophosphate (FdUMP) và 5-fluorouridine triphosphate (FUTP). Những chất chuyển hóa này sẽ làm tổn thương tế bào bằng hai cơ chế. Đầu tiên, FdUMP và đồng yếu tố folate N5-10-methylenetetrahydrofolate gắn với thymidylate synthase (TS) tạo nên một phức hợp gồm ba yếu tố đồng hóa trị. Sự gắn kết này sẽ ức chế sự hình thành thymidylate từ uracil. Thymidylate là một tiền chất cần thiết của thymidine triphosphate, một chất cần thiết cho sự tổng hợp DNA, vì vậy sự thiếu hụt hợp chất này có thể ức chế sự phân chia tế bào. Thứ hai, các men sao chép nhân có thể kết hợp một cách nhầm lẫn FUTP thay vì uridine triphosphate (UTP) trong quá trình tổng hợp RNA. Lỗi chuyển hóa này có thể ảnh hưởng tới sự tổng hợp RNA và protein.
Cơ chế tác dụng:
Capecitabine là dẫn xuất fluoropyrimidine carbamate được điều chế để dùng đường uống, là thuốc độc tế bào được hoạt hóa bởi khối u và chọn lọc trên khối u.
Capecitabine không phải là chất độc tế bào trên in vitro. Tuy nhiên, trên in vivo, thuốc được biến đổi liên tiếp thành chất gốc độc tế bào là 5-fluorouracil (5-FU), chất này sẽ được chuyển hoá tiếp.
Sự hình thành 5-FU tại khối u nhờ xúc tác một cách tối ưu của yếu tố tạo mạch liên quan tới khối u là thymidine phosphorylase (dThdPase), nhờ đó làm giảm tối đa mức độ của mô lành với 5-FU trong cơ thể.
[XEM TẠI ĐÂY]
5.2. Dược động học:
Hấp thu
Sau khi uống, capecitabine được hấp thu nhanh chóng và rộng khắp, sau đó được chuyển hóa mạnh thành chất chuyển hóa 5′-deoxy-5-fluorocytidine (5′-DFCR) và 5’DFUR. Dùng cùng với thức ăn làm giảm tỉ lệ hấp thu capecitabine, nhưng chỉ ảnh hưởng rất ít tới diện tích dưới đường cong (AUC) của 5’DFUR và chất chuyển hóa tiếp theo của nó là 5-FU. Với liều 1250mg/m2 vào ngày thứ 14 sau khi ăn, nồng độ đỉnh huyết tương (Cmax tính bằng μg/mL) cho capecitabine, 5′-DFCR, 5′-DFUR, 5-FU và FBAL tương ứng là 4,47; 3,05; 12,1; 0,95 và 5,46. Thời gian để đạt tới nồng độ đỉnh huyết tương (Tmax tính bằng giờ) tương ứng là 1,50; 2,00; 2,00; 2,00 và 3,34. Giá trị AUC0-∞ tính bằng μg.giờ/mL tương ứng là 7,75; 7,24; 24,6; 2,03 và 36,3.
Phân bố
Gắn kết với protein
Các nghiên cứu huyết tương trên in-vitro đã chứng minh rằng tỉ lệ gắn kết protein của capecitabine, 5′-DFCR, 5′-DFUR và 5-FU lần lượt là 54%, 10%, 62% và 10%, chủ yếu gắn với albumin.
Chuyển hóa
Capecitabine được chuyển hóa đầu tiên bởi men carboxylesterase ở gan thành 5′-DFCR, chất này sau đó được chuyển thành 5′-DFUR bởi cytidine deaminase, là men tập trung chủ yếu ở gan và mô khối u.
Sự hình thành 5-FU xảy ra chủ yếu tại vị trí khối u bởi yếu tố tạo mạch có liên quan đến khối u là dThdPase, do đó làm giảm tối đa mức độ của mô lành với 5-FU trong cơ thể.
AUC huyết tương của 5-FU thấp hơn 6 đến 22 lần nồng độ sau khi truyền tĩnh mạch nhanh 5-FU (liều 600mg/m2). Các chất chuyển hóa của capecitabine chỉ trở nên có độc tính sau khi chuyển thành 5-FU và các chất đồng hóa của 5-FU (xem phần Cơ chế hoạt động).
5-FU được chuyển hóa tiếp thành các chất chuyển hóa không có hoạt tính dihydro-5-fluoruracil (FUH2), 5 fluoro-ureidopropionic acid (FUPA) và α-fluoro-β-alanine (FBAL) thông qua dihydropyrimidine dehydrogenase (DPD), chất này mang tính chuyển hóa chậm.
Đào thải
Thời gian bán thải (t1/2 tính bằng giờ) của capecitabine, 5′-DFCR, 5′-DFUR, 5-FU và FBAL tương ứng là 0,85; 1,11; 0,66; 0,76 và 3,23. Dược động học của capecitabine được đánh giá trên khoảng liều là 502-3514mg/m2/ngày. Các thông số của capecitabine, 5′-DFCR và 5′-DFUR được đo vào ngày đầu tiên và ngày 14 là như nhau. AUC của 5-FU là 30%-35% cao hơn vào ngày 14, nhưng không tăng lên sau đó (ngày 22). Tại liều điều trị, dược động học của capecitabine và các chất chuyển hóa của nó tỉ lệ với liều, trừ 5-FU.
Sau khi uống, các chất chuyển hóa của capecitabine được tìm thấy chủ yếu trong nước tiểu. 95,5% liều capecitabine được dùng tìm thấy trong nước tiểu. Bài tiết trong phân rất ít (2,6%). Chất chuyển hóa chính có trong nước tiểu là FBAL, chiếm 57% liều dùng. Khoảng 3% liều dùng được đào thải trong nước tiểu dưới dạng thuốc không đổi.
Chế độ điều trị kết hợp
Các nghiên cứu pha I đánh giá tác động của Xelocapec lên dược động học của hoặc docetaxel hoặc paclitaxel và ngược lại cho thấy Xelocapec không tác động lên dược động học của docetaxel hoặc paclitaxel (Cmax và AUC) và docetaxel hoặc paclitaxel cũng không tác động lên dược động học của 5′-DFUR (chất chuyển hóa quan trọng nhất của capecitabine).
Dược động học ở dân số đặc biệt
Phân tích dược động học dân số được tiến hành sau khi điều trị Xelocapec ở 505 bệnh nhân bị ung thư đại trực tràng ở liều 1250mg/m2 hai lần mỗi ngày. Giới tính, có hoặc không có di căn ở gan lúc ban đầu, đánh giá tổng trạng Karnofsky, bilirubin toàn phần, albumin huyết thanh, ASAT và ALAT không có tác động có ý nghĩa thống kê lên dược động học của 5′-DFUR, 5-FU và FBAL.
Bệnh nhân bị suy gan do di căn ở gan: Không ghi nhận tác động có ý nghĩa lâm sàng của capecitabine lên hoạt tính sinh học và dược động học trên những bệnh nhân ung thư có chức năng gan giảm từ nhẹ đến trung bình do di căn ở gan (xem phần Các hướng dẫn liều dùng đặc biệt).
Không có dữ liệu dược động học trên những bệnh nhân suy gan nặng.
Bệnh nhân bị suy thận: Dựa vào nghiên cứu dược động học trên những bệnh nhân ung thư bị suy thận mức độ từ nhẹ đến nặng, không thấy bằng chứng về sự tác động của độ thanh thải creatinine lên dược động học của thuốc nguyên thủy và 5-FU. Nghiên cứu cho thấy độ thanh thải creatinine có ảnh hưởng đến mức độ tiếp xúc toàn thân với 5′-DFUR (AUC tăng 35% khi độ thanh thải giảm 50%) và với FBAL (AUC tăng 114% khi độ thanh thải creatinine giảm khoảng 50%). FBAL là chất chuyển hóa không có hoạt tính chống tăng sinh; 5′-DFUR là tiền chất trực tiếp của 5-FU (xem phần Các hướng dẫn liều dùng đặc biệt).
Người già: Dựa vào phân tích dược động học dân số, bao gồm những bệnh nhân có khoảng tuổi rộng (từ 27 đến 86 tuổi) và bao gồm 234 (46%) bệnh nhân có tuổi 65 trở lên, cho thấy tuổi không ảnh hưởng đến dược động học của 5′-DFUR và 5-FU. AUC của FBAL tăng theo tuổi (tuổi tăng 20% làm AUC của FBAL tăng 15%). Sự tăng này có thể do thay đổi chức năng thận (xem phần Các hướng dẫn liều dùng đặc biệt, xem phần Dược động học ở số dân đặc biệt, phần bệnh nhân bị suy thận).
Chủng tộc: Dựa vào phân tích dược động học dân số của 455 bệnh nhân da trắng (90,1%), 22 bệnh nhân da đen (4,4%) và 28 bệnh nhân của các chủng tộc hoặc sắc tộc khác (5,5%), dược động học của những bệnh nhân chủng da đen không khác dược động học ở chủng người da trắng.
5.3 Giải thích:
Chưa có thông tin. Đang cập nhật.
5.4 Thay thế thuốc :
Chưa có thông tin. Đang cập nhật.
*Lưu ý:
Các thông tin về thuốc trên Pharmog.com chỉ mang tính chất tham khảo – Khi dùng thuốc cần tuyệt đối tuân theo theo hướng dẫn của Bác sĩ
Chúng tôi không chịu trách nhiệm về bất cứ hậu quả nào xảy ra do tự ý dùng thuốc dựa theo các thông tin trên Pharmog.com
6. Phần thông tin kèm theo của thuốc:
6.1. Danh mục tá dược:
Tablet core: anhydrous lactose,, croscarmellose sodium, hypromellose (3 mPa.s), microcrystalline cellulose, magnesium stearate.
Tablet coating: hypromellose, titanium dioxide (E171), yellow iron oxide (E172), red iron oxide (E172), talc.
6.2. Tương kỵ :
Không áp dụng.
6.3. Bảo quản:
Không bảo quản thuốc ở nhiệt độ trên 30oC, giữ thuốc trong bao bì gốc để tránh ẩm.
6.4. Thông tin khác :
An toàn tiền lâm sàng
Trong các nghiên cứu độc tính lặp lại liều, dùng capecitabine đường uống hàng ngày trên khỉ cái đuôi dài và chuột đã gây ra những tác dụng độc hại trên hệ thống tiêu hóa, bạch huyết và hệ tạo máu, điển hình khi dùng fluoropyrimidines. Những độc tính này có thể hồi phục. Độc tính trên da, đặc trưng bởi những thay đổi thoái hóa, đã được quan sát khi dùng capecitabine. Capecitabine không gây nhiễm độc gan và thần kinh trung ương. Độc tính tim mạch (ví dụ PR và khoảng QT kéo dài) đã phát hiện ở loài khỉ cái đuôi dài sau khi tiêm tĩnh mạch (100 mg/kg) nhưng không phải sau khi lặp đi lặp lại một liều uống (1379 mg/m2/ngày).
Một nghiên cứu 2 năm về tính sinh ung trên chuột cho thấy không có bằng chứng gây ung thư bởi capecitabine. Trong quá trình sinh sản tiêu chuẩn, sự suy giảm khả năng sinh sản đã được quan sát thấy ở chuột cái dùng capecitaine; tuy nhiên, ảnh hưởng này có thể được phục hồi sau một thời gian không dùng thuốc. Ngoài ra, trong một nghiên cứu 13 tuần, đã xảy ra teo và thoái hóa cơ quan sinh sản của chuột đực; tuy nhiên những ảnh hưởng này có thể hồi phục sau một thời gian không dùng thuốc.
Trong các nghiên cứu khả năng gây độc tính phôi thai và gây quái thai ở chuột, đã quan sát thấy sự tăng liều dùng trong tái hấp thu của bào thai và gây quái thai. Trên khỉ, sẩy thai và thai chết lưu đã được quan sát ở liều cao, nhưng không có bằng chứng gây quái thai.
Capecitabine không gây đột biến trong ống nghiệm với vi khuẩn (thử nghiệm Ames) hoặc các tế bào động vật có vú (Hamster Trung Quốc V79/HPRT gen đột biến). Tuy nhiên, tương tự như các chất tương tự nucleoside khác (ví dụ 5-FU), capecitabine là yếu tố gây đột biến trong tế bào lympho người (in vitro) và một kết quả dương tính xảy ra ở tủy xương chuột thử nghiệm vi nhân tủy (in vivo).
6.5 Tài liệu tham khảo:
Dược Thư Quốc Gia Việt Nam
Hoặc HDSD Thuốc.