Thuốc PERJETA là thuốc gì ? Dưới đây là nội dung tờ hướng dẫn sử dụng gốc của Thuốc PERJETA (Thông tin bao gồm liều dùng, cách dùng, chỉ định, chống chỉ định, thận trọng, dược lý…)
1. Tên hoạt chất và biệt dược:
Hoạt chất : Pertuzumab
Phân loại: Thuốc chống ung thư. Kháng thể đơn dòng
Nhóm pháp lý: Thuốc kê đơn ETC – (Ethical drugs, prescription drugs, Prescription only medicine)
Mã ATC (Anatomical Therapeutic Chemical): L01XC13.
Brand name: PERJETA.
Hãng sản xuất : F.Hoffmann-La Roche Ltd.
2. Dạng bào chế – Hàm lượng:
Dạng thuốc và hàm lượng
Dung dịch đậm đặc pha truyền (không chất bảo quản): lọ 14mL. Mỗi mL: Pertuzumab 30mg.
Thuốc tham khảo:
| PERJETA 420 mg/14ml | ||
| Mỗi ml dung dịch đậm đặc có chứa: | ||
| Pertuzumab | …………………………. | 30 mg |
| Tá dược | …………………………. | vừa đủ (Xem mục 6.1) |

3. Video by Pharmog:
[VIDEO DƯỢC LÝ]
————————————————
► Kịch Bản: PharmogTeam
► Youtube: https://www.youtube.com/c/pharmog
► Facebook: https://www.facebook.com/pharmog/
► Group : Hội những người mê dược lý
► Instagram : https://www.instagram.com/pharmogvn/
► Website: pharmog.com
4. Ứng dụng lâm sàng:
4.1. Chỉ định:
Ung thư vú di căn: Perjeta được chỉ định điều trị kết hợp với Herceptin và docetaxel cho bệnh nhân ung thư vú di căn hoặc ung thư vú tái phát tại chỗ không thể phẫu thuật được, có HER2 dương tính, chưa được điều trị với liệu pháp kháng HER2 hoặc hóa trị liệu đối với ung thư di căn.
Điều trị tân bổ trợ ung thư vú: Perjeta phối hợp với Herceptin và hóa trị để điều trị tân bổ trợ cho những bệnh nhân ung thư vú có HER2 dương tính, tiến triển tại chỗ, viêm hoặc ung thư vú giai đoạn sớm (hoặc đường kính khối u > 2cm, hoặc có hạch dương tính) như là một phần trong phác đồ điều trị hoàn chỉnh cho bệnh nhân ung thư vú giai đoạn sớm (xem phần Liều lượng và Cách dùng và Thử nghiệm lâm sàng).
4.2. Liều dùng – Cách dùng:
Cách dùng :
Hướng dẫn pha thuốc
Thuốc Pertuzumab chỉ được dùng liều đơn qua đường truyền tĩnh mạch.
Pertuzumab không có chất bảo quản kháng khuẩn. Do đó phải đảm bảo sự vô khuẩn của dung dịch pha truyền, và phải do nhân viên y tế chuẩn bị.
Thuốc Pertuzumab phải do nhân viên y tế chuẩn bị, sử dụng kỹ thuật tiệt trùng.
14 ml dung dịch cô đặc thuốc Pertuzumab nên được lấy từ lọ và pha loãng trong túi PVC hoặc polyolefin không PVC chứa 250 ml dịch truyền NaCl 0,9%. Không lấy nước muối ra khỏi túi dịch truyền.
Với liều ban đầu, cần dùng 2 lọ và sau khi pha loãng, nồng độ pertuzumab trong dung dịch là 3,0 mg/ml, với liều tiếp theo, cần dùng 1 lọ, và sau khi pha loãng, nồng độ pertuzumab trong dung dịch là 1,6 mg/ml.
Không được dùng dung dịch Dextrose (5%) (xem phần Tương kỵ).
Trộn bằng cách xoay ngược túi thuốc nhẹ nhàng để tránh tạo bọt.
Các thuốc dùng đường tiêm truyền phải được kiểm tra bằng mắt các cặn lắng và sự mất màu sắc trước khi dùng. Thuốc phải được dùng liền sau khi chuẩn bị (xem phần Bảo quản).
Liều dùng:
Bệnh nhân điều trị với Pertuzumab cần phải có khối u HER2 dương tính, được định nghĩa là có điểm 3+ theo IHC hoặc là tỷ lệ >2,0 theo ISH được đánh giá bằng một thử nghiệm đã được kiểm định.
Để đảm bảo kết quả chính xác và có tính lặp lại, các xét nghiệm cần được thực hiện trong phòng xét nghiệm chuyên khoa, nơi có thể đảm bảo sự xác thực của các quy trình xét nghiệm.
Để được hướng dẫn đầy đủ về việc thực hiện xét nghiệm và diễn giải xin vui lòng tham khảo tờ thông tin về xét nghiệm HER2 đã được kiểm định.
Nếu muốn chuyển qua một sản phẩm thuốc sinh học khác, cần sự đồng ý của bác sĩ điều trị.
Để tránh sai sót khi sử dụng thuốc, cần kiểm tra nhãn thuốc để bảo đảm thuốc được chuẩn bị và sử dụng đúng là Pertuzumab.
Pertuzumab phải được dùng dưới sự giám sát của bác sĩ có kinh nghiệm điều trị bệnh nhân ung thư.
Pertuzumab phải được pha loãng bởi nhân viên y tế và được tiêm truyền tĩnh mạch. Không được tiêm hoặc bơm tĩnh mạch.
Liều lượng Pertuzumab khi phối hợp với Herceptin và docetaxel
Liều khởi đầu của Pertuzumab được khuyến cáo là 840 mg tiêm truyền tĩnh mạch trong 60 phút, tiếp theo 3 tuần sau đó là liều 420 mg được tiêm truyền từ 30 đến 60 phút.
Khi sử dụng chung với Pertuzumab, Herceptin được khuyến nghị là nên truyền tĩnh mạch với liều khởi đầu là 8 mg/kg trong vòng 90 phút theo lịch trình mỗi 3 tuần, tiếp theo mỗi 3 tuần sau đó với liều 6 mg/kg trong vòng từ 30 đến 90 phút.
Khi sử dụng chung với Pertuzumab, khuyến nghị liều khởi đầu của docetaxel là 75 mg/m2. Có thể tăng liều lên đến 100 mg/m2 nếu liều khởi đầu được dung nạp tốt.
Các thuốc nên được dùng tuần tự. Pertuzumab và Herceptin có thể cho theo bất kỳ thứ tự nào. Khi bệnh nhân dùng docetaxel, thì liều docetaxel phải được dùng sau Pertuzumab và Herceptin. Cần theo dõi bệnh nhân 30-60 phút sau mỗi lần truyền Pertuzumab và trước khi bắt đầu bất cứ việc tiêm truyền nào tiếp theo với Herceptin hoặc docetaxel (xem phần Cảnh báo).
Điều trị tân bổ trợ ung thư vú
Pertuzumab, Herceptin và docetaxel nên được dùng như trên như một phần của một trong các phác đồ sau:
3 chu kì sau phác đồ FEC
4 chu kì trước phác đồ FEC
6 chu kì với carboplatin (không nên tăng liều docetaxel trên 75 mg/m2).
Sau khi phẫu thuật, bệnh nhân nên được điều trị bổ trợ bằng Herceptin cho đủ 1 năm.
Chưa có đủ bằng chứng khuyến cáo dùng đồng thời một loại anthracycline với Pertuzumab.
Tính an toàn của Pertuzumab như một phần trong phác đồ điều trị có doxorubicin chưa được xác lập.
Thời gian điều trị:
Ung thư vú di căn: Khuyến cáo bệnh nhân nên điều trị với Pertuzumab đến khi nào bệnh tiến triển hoặc bị độc tính không thể kiểm soát được.
Điều trị tân bổ trợ ung thư vú:
Khuyến cáo bệnh nhân nên điều trị với Pertuzumab từ 3 đến 6 chu kì phụ thuộc vào phác đồ điều trị được chọn lựa (xem phần Thử nghiệm lâm sàng).
Tính an toàn của việc dùng Pertuzumab hơn 6 chu kỳ cho điều trị tân bổ trợ ung thư vú chưa được xác lập.
Hoãn hoặc bỏ liều: Nếu thời gian giữa 2 lần truyền dưới 6 tuần, liều Pertuzumab 420 mg nên được cho càng sớm càng tốt. Đừng chờ đến lần truyền dự kiến tiếp theo. Nếu thời gian giữa 2 lần truyền là 6 tuần hoặc hơn, nên dùng lại liều Pertuzumab 840 mg khởi đầu truyền tĩnh mạch trong vòng 60 phút, sau đó liều 420 mg mỗi 3 tuần truyền tĩnh mạch trong 30-60 phút.
Điều chỉnh liều:
Nên ngưng Pertuzumab nếu ngưng điều trị Herceptin.
Nếu ngưng docetaxel, vẫn có thể tiếp tục điều trị với Pertuzumab và Herceptin đến khi nào bệnh tiến triển hoặc độc tính không thể xử trí được.
Không khuyến nghị giảm liều đối với Pertuzumab.
Đối với Herceptin khuyến nghị không nên giảm liều, xin xem thông tin ghi toa Herceptin.
Đối với chỉnh liều docetaxel và các hóa chất khác, xin xem thông tin ghi toa của các thuốc liên quan.
Phản ứng liên quan đến tiêm truyền: Giảm tốc độ truyền hoặc ngưng truyền Pertuzumab nếu bệnh nhân có phản ứng liên quan đến tiêm truyền.
Phản ứng quá mẫn/sốc phản vệ: Nên ngưng tức thì tiêm truyền nếu bệnh nhân bị phản ứng quá mẫn nghiêm trọng (xem phần Cảnh báo).
Rối loạn chức năng thất trái:
Tạm ngưng Pertuzumab và Herceptin trong ít nhất 3 tuần đối với bất cứ trường hợp nào dưới đây:
Khi phân suất tống máu thất trái (LVEF) giảm xuống dưới 40%
Khi phân suất tống máu thất trái (LVEF) là 40-45% kết hợp với giảm ≥ 10% điểm so với mức trước khi điều trị.
Pertuzumab và Herceptin có thể dùng lại nếu LVEF phục hồi lại đến > 45% hoặc 40-45% kết hợp với giảm < 10% điểm dưới mức trước khi điều trị.
Nếu sau khi lặp lại sự đánh giá trong khoảng 3 tuần, LVEF không được cải thiện, hoặc giảm thêm, nên ngưng Pertuzumab và Herceptin, trừ khi đánh giá lợi ích vượt hơn nguy cơ đối với bệnh nhân (xem phần Cảnh báo).
Giới hạn sử dụng liên quan đến an toàn
Tính an toàn của việc dùng Pertuzumab trong liệu trình có doxorubicin chưa được xác lập.
Tính an toàn của việc dùng Pertuzumab hơn 6 chu kỳ ở bệnh nhân ung thư vú giai đoạn đầu chưa được xác lập.
Hướng dẫn về liều lượng đặc biệt
Người cao tuổi: Không ghi nhận khác biệt về tính an toàn và hiệu quả của Pertuzumab giữa bệnh nhân người lớn ≥65 tuổi và <65 tuổi. Không cần chỉnh liều ở bệnh nhân cao tuổi.
Trẻ em: Tính an toàn và hiệu quả của Pertuzumab ở trẻ em và thiếu niên dưới 18 tuổi chưa được xác lập.
Suy giảm chức năng thận: Không cần chỉnh liều Pertuzumab ở bệnh nhân có suy thận nhẹ và vừa. Không có khuyến cáo nào về liều lượng cho bệnh nhân suy thận nặng vì chưa có đủ dữ liệu dược động học (xem phần Dược động học ở những dân số đặc biệt).
Suy chức năng gan: Tính an toàn và hiệu quả của Pertuzumab không được nghiên cứu trên bệnh nhân suy gan.
4.3. Chống chỉ định:
Chống chỉ định Pertuzumab ở bệnh nhân đã biết có tình trạng quá mẫn với pertuzumab hoặc bất cứ tá dược nào của thuốc.
4.4 Thận trọng:
Tổng quát: Nhằm cải thiện việc truy xuất nguồn gốc của sản phẩm thuốc sinh học, tên thương mại của thuốc sử dụng phải được ghi rõ trong hồ sơ của bệnh nhân.
Suy giảm chức năng thất trái
Đã có báo cáo thuốc ức chế hoạt động HER2 làm suy giảm phân suất tống máu thất trái, kể cả Pertuzumab. Trong nghiên cứu then chốt Phase III CLEOPATRA, ở các bệnh nhân ung thư vú di căn, Pertuzumab kết hợp với Herceptin và docetaxel không có liên quan đến sự tăng tỷ lệ rối loạn chức năng tâm thu thất trái có triệu chứng (LVD [suy tim sung huyết]) hoặc giảm phân suất tống máu thất trái (LVEF) so với giả dược khi kết hợp với Herceptin và docetaxel (xem phần Tác dụng ngoại ý). Tuy nhiên, bệnh nhân đã điều trị trước đó bằng anthracycline hoặc xạ trị trước đó ở vùng ngực có thể có nguy cơ giảm LVEF cao hơn.
Ở các bệnh nhân điều trị tân bổ trợ (NEOSPHERE), tần suất bệnh nhân có LVD ở nhóm điều trị bằng Pertuzumab cao hơn nhóm điều trị bằng Herceptin và docetaxel. Ở những bệnh nhân điều trị bằng Pertuzumab phối hợp với Herceptin và docetaxel, đã quan sát thấy có sự tăng tỷ lệ bệnh nhân bị giảm LVEF, tuy nhiên LVEF đã hồi phục ≥ 50% ở tất cả các bệnh nhân.
Pertuzumab chưa được nghiên cứu ở bệnh nhân: có LVEF ≤ 50% trước khi điều trị, ở người có tiền sử bị suy tim sung huyết trước đó; suy giảm LVEF đến < 50% trong thời gian điều trị trước đó với liệu pháp hỗ trợ bằng Herceptin; có tình trạng có thể đưa đến suy giảm chức năng thất trái như cao huyết áp không kiểm soát được, nhồi máu cơ tim gần đây, loạn nhịp thất nặng cần điều trị hoặc có những liều cộng dồn anthracycline trước đó đến > 360 mg/m2 doxorubicin hoặc tương đương.
Đánh giá LVEF trước khi khởi đầu điều trị với Pertuzumab và với khoảng cách thời gian đều đặn (ví dụ mỗi 3 tháng ở những bệnh nhân điều trị ung thư vú di căn và mỗi 6 tuần ở những bệnh nhân điều trị tân bổ trợ) trong thời gian điều trị để đảm bảo LVEF ở trong các giới hạn bình thường. Nếu LVEF < 40% hoặc 40-45% kèm theo giảm ≥ 10% điểm dưới mức trước khi điều trị, phải tạm ngưng Pertuzumab và Herceptin và đánh giá lại LVEF trong vòng 3 tuần. Nếu LVEF không cải thiện, hoặc giảm thêm sau đó, nên ngưng Pertuzumab và Herceptin, trừ khi lợi ích cho bệnh nhân nhiều hơn nguy cơ (xem phần Liều lượng và Cách dùng).
Độc tính trên phôi thai: Pertuzumab có thể gây nguy hiểm cho thai nhi khi được sử dụng cho phụ nữ mang thai. Điều trị khỉ cái đuôi dài đang mang thai với pertuzumab gây ra thiểu ối, chậm phát triển thận của thai nhi, và gây chết thai. Nếu dùng Pertuzumab trong thời kỳ mang thai, hoặc nếu bệnh nhân có thai trong khi đang dùng thuốc hoặc trong vòng 7 tháng sau liều cuối cùng của Pertuzumab kết hợp với trastuzumab, bệnh nhân cần được thông báo về những nguy hiểm có khả năng xày ra trên thai nhi [xem mục Sử dụng trên các đối tượng đặc biệt]. Cần xác nhận tình trạng mang thai trước khi bắt đầu điều trị với Pertuzumab. Tư vấn cho bệnh nhân về nguy cơ chết phôi-thai, dị tật bẩm sinh và sự cần thiết của việc tránh thai trong và sau khi điều trị. Tư vấn cho bệnh nhân cần liên lạc ngay lập tức với cán bộ y tế nếu nghi ngờ đang mang thai. Nếu bệnh nhân dùng Pertuzumab trong thời kỳ mang thai, hoặc nếu bệnh nhân có thai trong khi đang dùng thuốc hoặc trong vòng 7 tháng sau liều cuối cùng của Pertuzumab kết hợp với trastuzumab, cần ngay lập tức báo cáo với F. Hoffmann-La Roche Ltd., văn phòng đại diện tại thành phố Hồ Chí Minh theo số điện thoại +84 9 77 17 42 42 hoặc +84 9 72 82 93 55, Fax: +84 8 38 21 62 76. Khuyến khích những phụ nữ điều trị Pertuzumab kết hợp trastuzumab khi đang mang thai hoặc trong vòng 7 tháng trước khi thụ thai tham gia vào Mother Pregnancy Registry bằng cách liên hệ qua số điện thoại +84 9 77 17 42 42 hoặc +84 9 72 82 93 55 (xem thông tin tư vấn bệnh nhân). Theo dõi thiếu ối ở bệnh nhân mang thai khi đang điều trị với Pertuzumab. Nếu thiếu ối xảy ra, thực hiện các xét nghiệm trên thai phù hợp với tuổi thai và các tiêu chuẩn cộng đồng về chăm sóc. Hiệu quả của việc truyền dịch tĩnh mạch trong việc xử trí tình trạng thiếu ối do dùng Pertuzumab không rõ.
Phản ứng liên quan đến truyền dịch và phản ứng quá mẫn/ phản ứng phản vệ: Tiêm truyền Pertuzumab có liên quan đến phản ứng do tiêm truyền (xem phần Tác dụng ngoại ý). Trong nghiên cứu 1, phản ứng do tiêm truyền được định nghĩa là bất kỳ phản ứng nào trong số phản ứng quá mẫn, phản ứng phản vệ, phản ứng truyền cấp tính, hoặc hội chứng giải phóng cytokine xảy ra trong một lần truyền hoặc trong cùng một ngày. Liều khởi đầu của Pertuzumab đã được đưa ra một ngày trước khi dùng trastuzumab và docetaxel để cho phép đánh giá các phản ứng liên quan đến Pertuzumab. Vào ngày đầu tiên, khi chỉ Pertuzumab được dùng, tần suất chung của phản ứng do tiêm truyền là 13,0% ở nhóm điều trị với Pertuzumab và 9,8% ở nhóm dùng placebo. Có ít hơn 1% ở độ 3 hay 4. Các phản ứng liên quan đến tiêm truyền thường gặp nhất (≥ 1,0%) là sốt, ớn lạnh, mệt mỏi, nhức đầu, suy nhược, quá mẫn, và ói mửa. Trong liệu trình thứ hai khi tất cả các loại thuốc được tiêm vào cùng một ngày, các phản ứng liên quan đến tiêm truyền thường gặp nhất trong nhóm điều trị với Pertuzumab (≥ 1,0%) là mệt mỏi, rối loạn vị giác, mẫn cảm, đau cơ, và ói mửa. Trong nghiên cứu 2 và 3, Pertuzumab được tiêm vào cùng một ngày với các loại thuốc khác. Các phản ứng do tiêm truyền cũng phù hợp với những kết quả quan sát được trong nghiên cứu 1, với đa số phản ứng được ghi nhận là độ 1-2 theo Các tiêu chuẩn chung cho các phản ứng bất lợi của Viện Ung thư Quốc gia (NCI – CTCAE v3.0). Sau khi tiêm truyền lần đầu Pertuzumab, nên theo dõi sát bệnh nhân trong vòng 60 phút, và trong vòng 30 phút sau những lần tiêm truyền Pertuzumab tiếp theo. Nếu có phản ứng đáng kể do tiêm truyền xảy ra thì giảm tốc độ truyền hoặc ngưng lại và điều trị nội khoa thích hợp. Bệnh nhân cần được đánh giá lại và theo dõi sát đến khi nào tất cả triệu chứng biến mất. Cần xem xét việc ngưng truyền thuốc vĩnh viễn ở bệnh nhân bị phản ứng do tiêm truyền nặng. Việc đánh giá lâm sàng này phải dựa trên mức độ nặng của phản ứng xảy ra trước đó và đáp ứng đối với điều trị phản ứng bất lợi (xem phần Liều lượng và Cách dùng).
Phản ứng quá mẫn/sốc phản vệ:
Trong nghiên cứu 1, tần suất chung của phản ứng quá mẫn/sốc phản vệ là 10,8% ở nhóm bệnh nhân điều trị bởi Pertuzumab và 9,1% ở nhóm dùng placebo. Tỉ lệ các phản ứng quá mẫn/sốc phản vệ độ 3, 4 là 2,0% ở nhóm được điều trị bởi Pertuzumab và 2,5% ở nhóm dùng placebo theo NCI – CTCAE v3.0. Kết quả ghi nhận có 4 bệnh nhân trong nhóm được điều trị bởi Pertuzumab và 2 bệnh nhân ở nhóm dùng placebo có sốc phản vệ. Trong nghiên cứu 2 và 3, các phản ứng quá mẫn/sốc phản vệ phù hợp với kết quả quan sát được trong nghiên cứu 1. Trong nghiên cứu 2, hai bệnh nhân điều trị với Pertuzumab và docetaxel bị sốc phản vệ. Trong nghiên cứu 3, tần suất chung của phản ứng quá mẫn/sốc phản vệ cao nhất trong nhóm điều trị bởi Pertuzumab kết hợp TCH (13,2%), trong đó 2,6% là NCI-CTCAE (phiên bản 3) độ 3-4.
Bệnh nhân nên được theo dõi chặt chẽ để nhận diện các phản ứng quá mẫn. Phản ứng quá mẫn nghiêm trọng, bao gồm sốc phản vệ đã được quan sát thấy ở các thử nghiêm lâm sàng có điều trị bằng Pertuzumab (xem phần Tác dụng ngoại ý). Các thuốc điều trị những phản ứng như vậy, cũng như các thiết bị cấp cứu, cần được có sẵn để dùng ngay lập tức. Chống chỉ định dùng Pertuzumab với ở những bệnh nhân phản ứng quá mẫn với pertuzumab hoặc bất kì thành phần nào của thuốc đã được ghi nhận (xem phần Chống chỉ định).
Xét nghiệm HER2
Phát hiện HER2 biểu hiện quá mức là cần thiết để lựa chọn bệnh nhân thích hợp điều trị với Pertuzumab vì đây là những đối tượng duy nhất được nghiên cứu và được chứng minh có lợi ích [xem Chỉ định và sử dụng (1) và các nghiên cứu lâm sàng]. Bệnh nhân ung thư vú cần phải có bằng chứng HER2 biểu hiện quá mức được định nghĩa là có điểm 3+ theo IHC hoặc là tỷ lệ khuếch đại FISH ≥ 2.0 trong các nghiên cứu lâm sàng (FISH: Phương pháp xác định trình trạng HER2 bằng kỹ thuật lai tại chỗ gắn huỳnh quang). Chỉ có các dữ liệu hạn chế đối với bệnh nhân ung thư vú dương tính với FISH, nhưng không có protein biểu hiện quá mức trên IHC.
Đánh giá về tình trạng HER2 nên được thực hiện tại các phòng thí nghiệm bằng các xét nghiệm công nghệ cao đã được FDA chấp thuận. Việc thực hiện khảo nghiệm không đúng cách, bao gồm sử dụng mô được cố định chưa tối ưu, việc không sử dụng thuốc thử đặc biệt, độ lệch kết quả từ các hướng dẫn xét nghiệm cụ thể, và không có các kiểm tra thích hợp để chuẩn hóa xét nghiệm, có thể dẫn đến kết quả không đáng tin cậy.
Sử dụng ở trẻ em: Tính an toàn và hiệu quả của Pertuzumab ở trẻ em và thiếu niên dưới 18 tuổi chưa được xác lập.
Suy chức năng thận: (xem phần Hướng dẫn đặc biệt về liều lượng, Dược động học ở những dân số đặc biệt).
Suy chức năng gan: Tính an toàn và hiệu quả của Pertuzumab chưa được nghiên cứu ở những bệnh nhân suy gan.
Tác động của thuốc trên người lái xe và vận hành máy móc.
Khả năng lái xe và vận hành máy móc: Không có nghiên cứu nào về tác dụng của thuốc trên khả năng lái xe và vận hành máy móc được thực hiện.
Cần thận trọng khi sử dụng cho các đối tượng lái xe và vận hành máy móc.
4.5 Sử dụng cho phụ nữ có thai và cho con bú:
Xếp hạng cảnh báo
AU TGA pregnancy category: D
US FDA pregnancy category: NA
Thời kỳ mang thai:
Không nên sử dụng Pertuzumab ở phụ nữ có thai trừ khi lợi ích tiềm tàng cho mẹ vượt hơn nguy cơ có thể xảy ra cho bào thai. Phụ nữ trong tuổi có thể mang thai và bạn tình của những nam bệnh nhân, có khả năng có thai nên sử dụng biện pháp tránh thai hữu hiệu khi được điều trị với Pertuzumab và trong vòng 6 tháng sau liều cuối cùng Pertuzumab.
Không có nghiên cứu nào về Pertuzumab trên phụ nữ có thai. Khi tiêm Pertuzumab ở khỉ cynomolgus trong thời gian tượng hình thai thấy gây thiểu ối, làm chậm phát triển thận và gây tử vong cho bào thai (xem phần Tính sinh quái thai).
Chuyển dạ và sinh đẻ: Việc sử dụng an toàn Pertuzumab trong thời gian chuyển dạ và sinh đẻ chưa được thiết lập.
Thời kỳ cho con bú:
Vì lý do IgG người được tiết qua sữa mẹ và khả năng hấp thu, gây hại cho trẻ nhũ nhi không được biết, nên cần quyết định ngưng cho bú, hoặc cần xem xét tầm quan trọng của thuốc đối với mẹ và thời gian bán hủy của pertuzumab (xem phần Thải trừ ).
4.6 Tác dụng không mong muốn (ADR):
Thử nghiệm lâm sàng: Tính an toàn của Pertuzumab được đánh giá trên 600 bệnh nhân trong nghiên cứu ngẫu nhiên CLEOPATRA (n=808), nghiên cứu NEOSPHERE (n=417) và TRYPHAENA (n=225) và trong nghiên cứu pha I và II thực hiện trên bệnh nhân có những bệnh ác tính khác nhau, và điều trị chủ yếu với Pertuzumab kết hợp với các thuốc kháng ung thư khác. Tính an toàn của Pertuzumab qua các nghiên cứu pha I và II nói chung đều nhất quán với các kết quả quan sát được trong thử nghiệm CLEOPATRA, NEOSPHERE và TRYPHAENA, mặc dù tần suất và các phản ứng bất lợi hay gặp nhất thay đổi tùy thuộc vào việc sử dụng Pertuzumab đơn trị liệu hay kết hợp chung với các thuốc kháng sự tân sinh khác.
Ung thư vú di căn: Trong nghiên cứu ung thư vú di căn (MBC) then chốt CLEOPATRA, tác dụng không mong muốn thường gặp nhất (≥ 50%) được quan sát thấy khi dùng Pertuzumab kết hợp với Herceptin và docetaxel gồm tiêu chảy, rụng tóc, và giảm bạch cầu đa nhân trung tính. Tác dụng phụ độ 3-4 theo NCI-CTCAE (phiên bản 3) hay gặp nhất (≥ 10%) là giảm bạch cầu đa nhân trung tính, giảm bạch cầu trung tính có sốt và giảm bạch cầu. Sau khi ngưng dùng docetaxel, ADRs trong nhóm điều trị bằng Pertuzumab và Herceptin xảy ra với tỷ lệ < 10% bệnh nhân ngoại trừ tiêu chảy (28,1%), phát ban (18,3%), nhiễm trùng đường hô hấp trên (18,3%), đau đầu (17,0%), viêm mũi họng (17,0%), ngứa (13,7%), mệt mỏi (13,4%), suy nhược (13,4%), buồn nôn (12,7%) và đau khớp (11,4%).
Điều trị tân bổ trợ ung thư vú:
Trong nghiên cứu NEOSPHERE, các tác dụng không mong muốn (ADRs) phổ biến nhất (≥ 50%) được quan sát thấy khi dùng Pertuzumab kết hợp với Herceptin và docetaxel là rụng tóc và giảm bạch cầu đa nhân trung tính. ADR độ 3-4 phổ biến nhất theo NCI-CTCAE (phiên bản 3) (≥ 10%) là giảm bạch cầu đa nhân trung tính.
Trong nghiên cứu TRYPHAENA, khi Pertuzumab được dùng kết hợp với Herceptin và docetaxel trong ba chu kỳ sau ba chu kỳ FEC, ADRs phổ biến nhất (≥ 50%) là tiêu chảy, buồn nôn và rụng tóc. ADR độ 3-4 phổ biến nhất theo NCI-CTCAE (phiên bản 3) (≥ 10%) là giảm bạch cầu đa nhân trung tính và giảm bạch cầu. Tương tự, khi Pertuzumab được dùng kết hợp với docetaxel, carboplatin và Herceptin (TCH) trong sáu chu kỳ, các ADRs phổ biến nhất (≥ 50%) là tiêu chảy và rụng tóc. ADR độ 3-4 phổ biến nhất theo NCI-CTCAE (phiên bản 3) (≥ 10%) là giảm bạch cầu đa nhân trung tính, sốt do giảm bạch cầu đa nhân trung tính, thiếu máu, giảm bạch cầu và tiêu chảy.
Ung thư vú di căn và điều trị tân bổ trợ ung thư vú: Bảng 4 tóm tắt các ADRs của nhóm điều trị trong nghiên cứu mù đôi then chốt, CLEOPATRA, trong đó Pertuzumab kết hợp với Herceptin và docetaxel ở những bệnh nhân ung thư vú di căn (MBC), và của những nghiên cứu điều trị tân bổ trợ NEOSPHERE và TRYPHAENA, trong đó Pertuzumab kết hợp với Herceptin và hóa trị cho bệnh nhân tái phát tại chỗ, viêm hoặc ung thư vú giai đoạn sớm. Vì Pertuzumab được sử dụng với Herceptin và hóa trị, rất khó để xác định mối quan hệ nhân quả của một phản ứng bất lợi cho một loại thuốc riêng biệt.
Mức độ xảy ra các tác dụng không mong muốn được phân loại như sau: rất thường gặp (≥1/10), thường gặp (từ ≥1/100 đến <1/10), ít gặp (từ ≥1/1000 đến <1/100), hiếm (từ ≥1/10.000 để <1/1000), rất hiếm (từ <1/10.000).
| Bảng 4: Tóm tắt các phản ứng bất lợi xảy ra khi điều trị với Pertuzumab ờ các bệnh nhân di căn và điều trị tân bổ trợ | |||
| Phản ứng bất lợi (MedDRA) Hệ thống cơ quan | Pertuzumab + Herceptin + hóa trịAA n = 980AAA (100%) Tần suất % |
Loại tần suất | |
| Tất cả mức độ % | Độ 3-4% | ||
| Rối loạn huyết học và hệ bạch huyết | |||
| Giảm bạch cầu đa nhân trung tính | 45,2 | 41,2 | Rất thường gặp |
| Thiếu máu | 17,1 | 3,1 | Rất thường gặp |
| Giảm bạch cầu | 14,2 | 9,7 | Rất thường gặp |
| Sốt giảm bạch cầu* | 10,7 | 10,4 | Rất thường gặp |
| Rối loạn tim mạch | |||
| Rối loạn chức năng thất trái** | 4,5 | 0,8 | Thường gặp |
| Suy tim sung huyết** | 0,3 | 0,2 | ít gặp |
| Rối loạn về mắt | |||
| Tăng tiết nước mắt | 9 | – | Thường gặp |
| Rối loạn dạ dày – ruột | |||
| Tiêu chảy | 58,9 | 6,6 | Rất thường gặp |
| Buồn nõn | 39,5 | 0,7 | Rất thường gặp |
| Nôn | 23,4 | 1,3 | Rất thường gặp |
| Viêm dạ dày | 15,2 | 0,2 | Rất thường gặp |
| Táo bón | 12,3 | – | Rất thường gặp |
| Rối loạn toàn thân và tinh trạng tại vị trí tiêm | |||
| Mệt mỏi | 31,5 | 1,4 | Rất thường gặp |
| Viêm niêm mạc | 21,5 | 0,9 | Rất thường gặp |
| Suy nhược | 18,6 | 1,7 | Rất thường gặp |
| Sốt | 15,6 | 0,5 | Rất thường gặp |
| Phù ngoại vi | 12,4 | 0,2 | Rất thường gặp |
| Rối loạn hệ thống miễn dịch | |||
| Quá mẫn với thuốc | 5,2 | 0,9 | Thường gặp |
| Quá mẫn | 3,1 | 0,4 | Thường gặp |
| Nhiễm khuẩn và nhiễm kỷ sinh trùng | |||
| Nhiễm khuẩn đường hô hấp trẽn | 11,7 | 0,3 | Rất thường gặp |
| Viêm mũi hầu | 10,8 | – | Rất thường gặp |
| Viêm quanh móng | 3,8 | – | Thường gặp |
| Rối loạn chuyển hóa và dinh dưỡng | |||
| Giảm thèm ăn | 19,6 | 0,7 | Rất thường gặp |
| Rối loạn cơ – xương, mõ liên kết | |||
| Đau cơ | 18,6 | 0,6 | Rất thường gặp |
| Đau khớp | 13,2 | 0,1 | Rất thường gặp |
| Rối loạn hệ thần kinh | |||
| Đau đầu | 19,3 | 0,7 | Rất thường gặp |
| Rối loạn vị giác | 14,1 | – | Rất thường gặp |
| Bệnh lý thần kinh ngoại vi | 11,6 | 1,1 | Rất thường gặp |
| Chóng mặt | 9,9 | 0,4 | Thường gặp |
| Bệnh lý thần kinh cảm giác ngoại vi | 8,9 | 0,3 | Thường gặp |
| Rối loạn tâm thần | |||
| Mất ngủ | 12,2 | – | Rất thường gặp |
| Rối loạn hô hấp, lồng ngực và trung thất | |||
| Khó thở | 9,8 | 0,7 | Thường gặp |
| Tràn dịch màng phổi | 2,2 | 0,1 | Thường gặp |
| Rối loạn về da và mô dưới da | |||
| Rụng tóc | 51 | – | Rất thường gặp |
| Phát ban | 27,1 | 0,6 | Rất thường gặp |
| Rối loạn về móng | 13,5 | 0,5 | Rất thường gặp |
| Ngứa | 9,2 | – | Thường gặp |
| Khô da | 7,7 | – | Thường gặp |
A Bảng 4 trình bày dữ liệu từ giai đoạn điều trị tổng thể trong nghiên cứu CLEOPATRA (dữ liệu lấy tại thời điểm ngày 11/02/014, số chu kỳ điều trị trung bình của Pertuzumab là 24); và từ thời gian điều trị tân bổ trợ trong nghiên cứu NEOSPHERE (số chu kỳ điều trị trung bình của Pertuzumab là 4, trẽn tất cả các nhóm điều trị) và nghiên cứu TRYPHAENA(số chu kỳ điều trị trung bình của Pertuzumab là 3 trong nhóm FEC / Ptz + T + D và 6 trong nhóm Ptz + T + FEC / Ptz + T + D và Ptz + TCH).
AA Trong nghiên cứu NEOSPHERE, 108 bệnh nhân điều trị với Pertuzumab + Herceptin mà không kết hợp với docetaxel và 94 bệnh nhân điều trị với Pertuzumab + docetaxel mà không kết hợp với Herceptin.
AAA Trong nghiên cứu CLEOPATRA, trang tổng số 980 bệnh nhân điều trị với Parjeta, 45 bệnh nhân được chọn ngẫu nhiên điều trị với giả dược không có tiền sử dùng Pertuzumab, đã chuyển sang dùng Pertuzumab.
* Bảng này trình bày một phản ứng bất lợi đã được báo cáo có liên quan với hậu quả gây tử vong.
** Tỷ lệ rối loạn chức năng thất trái và suy tim sung huyết phản ánh các phản ứng bất lợi được báo cáo trong các nghiên cứu riêng biệt.
Thông tin thêm về các phản ứng bất lợi chọn lọc:
Rối loạn chức năng thất trái (LVD)
Trong thử nghiệm then chốt CLEOPATRA, tỷ lệ bệnh nhân bị LVD trong suốt quá trình điều trị ở nhóm dùng giả dược cao hơn nhóm dùng Pertuzumab (8,6% và 6,6%, theo thứ tự). Tỷ lệ bệnh nhân bị LVD có biểu hiện triệu chứng cũng thấp hơn ở nhóm dùng Pertuzumab (1,8% ở nhóm giả dược với 1,5% nhóm điều trị với Pertuzumab) (xem phần Cảnh báo).
Trong nghiên cứu NEOSPHERE, những bệnh nhân dùng 4 chu kì tân bổ trợ Pertuzumab, có tỷ lệ LVD (trong suốt quá trình điều trị) ở nhóm dùng Pertuzumab, Herceptin và docetaxel cao hơn (7,5%) so với nhóm dùng Herceptin và docetaxel (1,9%). Có 1 trường hợp bệnh nhân có triệu chứng LVD ở nhóm dùng Pertuzumab và Herceptin.
Trong nghiên cứu TRYPHAENA, tỷ lệ bệnh nhân bị LVD (trong suốt quá trình điều trị) là 8,3% ở nhóm dùng Pertuzumab, Herceptin và FEC theo sau đó dùng Pertuzumab, Herceptin và docetaxel; 9,3% ở nhóm bệnh nhân dùng FEC theo sau đó dùng Pertuzumab, Herceptin và Docetaxel; 6,6% ở nhóm bệnh nhân dùng Pertuzumab phối hợp với TCH. Tỷ lệ bệnh nhân có triệu chứng LVD (suy tim sung huyết) là 1,3% ở nhóm dùng FEC theo sau đó dùng Pertuzumab, Herceptin và docetaxel (đã loại trừ một bệnh nhân có triệu chứng LVD trong quá trình điều trị với FEC trước khi điều trị Pertuzumab, Herceptin và docetaxel) và 1,3% ở nhóm điều trị với Pertuzumab kết hợp với TCH. Không có bệnh nhân trong nhóm điều trị bằng Pertuzumab, Herceptin và FEC theo sau đó dùng Pertuzumab, Herceptin và docetaxel có triệu chứng LVD.
Phản ứng liên quan đến tiêm truyền
Trong nghiên cứu then chốt pha III CLEOPATRA, phản ứng liên quan đến tiêm truyền được định nghĩa là bất kỳ biến cố nào như quá mẫn, phản ứng phản vệ, phản ứng cấp tính do tiêm truyền hoặc hội chứng giải phóng cytokine xảy ra trong thời gian tiêm truyền hoặc trong cùng ngày tiêm truyền. Trong nghiên cứu then chốt CLEOPATRA, liều đầu tiên của Pertuzumab được cho vào ngày trước khi điều trị với Herceptin và docetaxel để quan sát theo dõi các phản ứng liên quan đến Pertuzumab. Vào ngày đầu tiên khi chỉ truyền Pertuzumab, tần suất chung của các phản ứng liên quan đến tiêm truyền là 9,8% trong nhóm điều trị với giả dược và 13,2% trong nhóm điều trị với Pertuzumab, đa số các phản ứng là nhẹ và vừa. Phản ứng liên quan đến tiêm truyền thường xảy ra nhiều nhất (≥ 1,0%) trong nhóm Pertuzumab là sốt, rét run, mệt mỏi, đau đầu, suy nhược, quá mẫn và nôn.
Trong chu kỳ thứ hai, khi tất cả các thuốc được truyền cùng một ngày, phản ứng liên quan đến tiêm truyền xảy ra nhiều nhất (≥ 1,0%) trong nhóm điều trị với Pertuzumab là mệt mỏi quá mẫn với thuốc, rối loạn vị giác, quá mẫn, đau cơ và nôn (xem phần Cảnh báo).
Trong nghiên cứu NEOSPHERE và TRYPHAENA, Pertuzumab được dùng cùng một ngày như các loại thuốc điều trị khác. Các phản ứng do tiêm truyền cũng phù hợp với các phản ứng quan sát được trong nghiên cứu CLEOPATRA, với mức độ nghiêm trọng của các phản ứng là nhẹ hoặc trung bình.
Phản ứng quá mẫn/phản ứng phản vệ
Trong nghiên cứu then chốt CLEOPATRA tần suất chung các phản ứng quá mẫn/phản vệ được báo cáo là 9,3% trong nhóm điều trị với giả dược và 11,3% trong nhóm điều trị với Pertuzumab, trong đó độ 3-4 theo NCI-CTCAE (phiên bản 3), lần lượt là 2,5% và 2,0% theo thứ tự đó. Tính chung, 2 bệnh nhân trong nhóm điều trị với giả dược và 4 bệnh nhân trong nhóm điều trị với Pertuzumab bị phản ứng phản vệ (xem phần Cảnh báo).
Nói chung, đa số các phản ứng quá mẫn có mức độ nhẹ hay vừa đã được giải quyết qua điều trị. Dựa vào các thay đổi trong điều trị nghiên cứu, đa số các phản ứng đều được đánh giá là thứ phát sau khi truyền docetaxel.
Trong nghiên cứu NEOSPHERE và TRYPHAENA, các biến cố quá mẫn/phản ứng phản vệ phù hợp với những biến cố quan sát được trong nghiên cứu CLEOPATRA. Trong nghiên cứu NEOSPHERE, hai bệnh nhân trong nhóm dùng Pertuzumab và docetaxel bị phản ứng phản vệ. Trong nghiên cứu TRYPHAENA, tỷ lệ toàn bộ các phản ứng quá mẫn/phản ứng phản vệ là cao nhất ở nhóm dùng Pertuzumab và TCH (13,2%), trong đó 2,6% là độ 3-4 theo NCI-CTCAE (phiên bản 3).
Các bất thường về xét nghiệm: Trong nghiên cứu then chốt CLEOPATRA, tỉ lệ các biến cố bất lợi độ 3-4 theo NCI-CTCAE (phiên bản 3) về giảm số lượng bạch cầu trung tính tương tự như nhau trong 2 nhóm điều trị.
Hậu tiếp thị:
Báo cáo an toàn từ các báo cáo hậu tiếp thị phù hợp với dữ liệu an toàn từ các thử nghiệm lâm sàng của Pertuzumab.
Các bất thường xét nghiệm: Các bất thường xét nghiệm từ các báo cáo hậu tiếp thị phù hợp với dữ liệu từ các thử nghiệm lâm sàng của Pertuzumab.
Thông báo cho Bác sĩ những tác dụng không mong muốn gặp phải khi sử dụng thuốc.
4.7 Hướng dẫn cách xử trí ADR:
Ngừng sử dụng thuốc. Với các phản ứng bất lợi nhẹ, thường chỉ cần ngừng thuốc. Trường hợp mẫn cảm nặng hoặc phản ứng dị ứng, cần tiến hành điều trị hỗ trợ (giữ thoáng khí và dùng epinephrin, thở oxygen, dùng kháng histamin, corticoid…).
4.8 Tương tác với các thuốc khác:
Một nghiên cứu phụ trên 37 bệnh nhân của nghiên cứu then chốt CLEOPATRA, cho thấy không có bằng chứng về tương tác giữa Pertuzumab và Herceptin và giữa Pertuzumab và docetaxel. Ngoài ra không có bằng chứng tương tác lâm sàng nào, liên quan đến dược động học của Pertuzumab khi sử dụng chung với docetaxel hoặc Herceptin dựa trên phân tích dược động học dân số. Điều này đã được chứng minh bằng các dữ liệu dược động học trong nghiên cứu NEOSPHERE.
Bốn nghiên cứu được thực hiện để đánh giá tác dụng của Pertuzumab trên dược động học khi dùng chung với các chất độc tế bào khác, docetaxel, gemcitabine, erlotinib và capecitabine. Không có bằng chứng nào về tương tác dược động học giữa Pertuzumab và các thuốc đó. Dược động học của Pertuzumab trong các nghiên cứu này cũng tương tự như kết quả trong các nghiên cứu đơn trị liệu.
4.9 Quá liều và xử trí:
Không có kinh nghiệm về quá liều trong các thử nghiệm lâm sàng trên người. Các liều đơn cao hơn 25 mg/kg (1727 mg) chưa từng được thử nghiệm.
5. Cơ chế tác dụng của thuốc :
5.1. Dược lực học:
Các nghiên cứu lâm sàng/hiệu quả
Ung thư vú di căn
Pertuzumab phối hợp với Herceptin và docetaxel.
CLEOPATRA là một nghiên cứu lâm sàng pha III, đa trung tâm, ngẫu nhiên, mù đôi, đối chứng giả dược thực hiện trên 808 bệnh nhân bị ung thư vú di căn hoặc ung thư vú tái phát tại chỗ không thể phẫu thuật cắt bỏ có HER2 dương tính, chưa được điều trị với phác đồ kháng HER2 trước đó hoặc hóa trị liệu cho bệnh di căn. Mẫu bệnh phẩm khối u vú phải biểu hiện HER2 quá mức theo định nghĩa là IHC 3+ hoặc tỉ lệ khuếch đại ISH ≥ 2,0 được xác định bởi phòng xét nghiệm trung tâm. Các bệnh nhân được phân ngẫu nhiên theo tỉ lệ 1:1 vào nhóm điều trị giả dược cộng Herceptin và docetaxel, hoặc Pertuzumab cộng Herceptin và docetaxel. Việc phân nhóm ngẫu nhiên được phân tầng theo tình trạng điều trị trước đó (điều trị lần đầu hoặc đã điều trị hỗ trợ/tân hỗ trợ trước đó) và theo vùng địa lý (Châu Âu, Bắc Mỹ, Nam Mỹ và Châu Á). Những bệnh nhân đã được điều trị hỗ trợ hoặc tân hỗ trợ trước đó được yêu cầu có thời gian không bệnh ít nhất 12 tháng trước khi tuyển chọn vào nghiên cứu.
Pertuzumab được truyền tĩnh mạch liều ban đầu 840 mg, sau đó mỗi ba tuần với liều 420 mg. Herceptin được truyền tĩnh mạch với liều ban đầu 8 mg/kg, sau đó mỗi ba tuần với liều 6 mg/kg. Các bệnh nhân được điều trị với Pertuzumab và Herceptin cho tới khi bệnh tiến triển, đến khi bệnh nhân rút lui không tham gia nữa, hoặc tới khi xảy ra độc tính không xử trí được. Docetaxel được dùng với liều đầu tiên 75 mg/m2 truyền tĩnh mạch mỗi 3 tuần với ít nhất 6 chu kỳ. Liều docetaxel có thể tăng lên 100 mg/m2 tùy theo đánh giá của nghiên cứu viên nếu liều ban đầu được dung nạp tốt.
Tại thời điểm phân tích sơ khởi, số lượng chu kỳ điều trị thuốc nghiên cứu trung bình cho bệnh nhân là 16,2 trong nhóm dùng giả dược và 19,9 trong nhóm điều trị Pertuzumab.
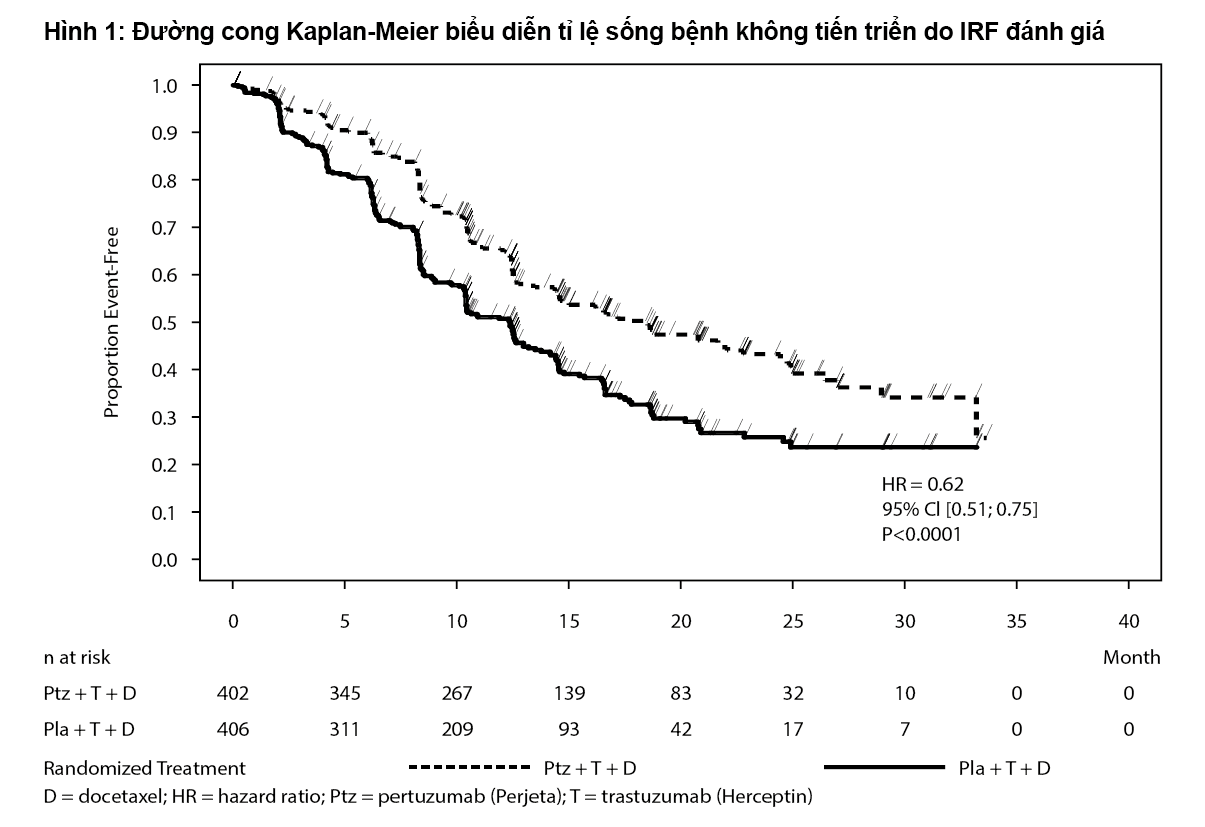
Tiêu chí chính của nghiên cứu là tỉ lệ sống bệnh không tiến triển (PFS) dựa trên đánh giá của hội đồng đánh giá độc lập (IRF) và được định nghĩa là thời gian từ ngày phân nhóm ngẫu nhiên đến ngày bệnh tiến triển hoặc tử vong (do mọi nguyên nhân) nếu tử vong xảy ra trong vòng 18 tuần từ lần đánh giá khối u sau cùng. Các tiêu chí phụ là tỉ lệ sống còn toàn bộ (OS), PFS (theo đánh giá của nghiên cứu viên), tỉ lệ đáp ứng chung (ORR), thời gian đáp ứng, và thời gian đến khi triệu chứng tiến triển theo bảng câu hỏi FACT B QoL.
Đặc điểm nhân chủng học được cân bằng tốt (tuổi trung bình 54, đa số là người da trắng (59%) và tất cả là phụ nữ, ngoại trừ hai bệnh nhân). Khoảng một phần hai bệnh nhân trong mỗi nhóm điều trị có thụ thể hormone dương tính (được định nghĩa là thụ thể estrogen [ER] dương tính và/hoặc thụ thể progesterone [PgR] dương tính) và khoảng một phần hai bệnh nhân trong mỗi nhóm đã điều trị hỗ trợ (adjuvant) hoặc tân hỗ trợ (neoadjuvant) trước đó (192 bệnh nhân [47,3%] trong nhóm dùng giả dược so với 184 bệnh nhân [45,8%] trong nhóm điều trị Pertuzumab).
Tại thời điểm phân tích sơ khởi về tỉ lệ sống bệnh không tiến triển, tổng cộng có 242 bệnh nhân (59%) trong nhóm dùng giả dược và 191 bệnh nhân (47,5%) trong nhóm điều trị với Pertuzumab có bệnh tiến triển do IRF xác định hoặc tử vong trong vòng 18 tuần từ ngày đánh giá khối u lần cuối cùng.
Tại thời điểm phân tích ban đầu, nghiên cứu CLEOPATRA đã chứng minh sự cải thiện có ý nghĩa thống kê về PFS do IRF đánh giá (tỉ số nguy cơ [HR]=0,62, 95% CI=0,51, 0,75, p<0,0001) ở nhóm điều trị Pertuzumab so với nhóm giả dược, và sự gia tăng trung vị PFS 6,1 tháng (PFS trung bình 12,4 tháng trong nhóm giả dược so với 18,5 tháng trong nhóm điều trị Pertuzumab). Các kết quả PFS đánh giá bởi nghiên cứu viên tương đồng với những kết quả quan sát thấy khi PFS được IRF đánh giá (PFS trung bình là 12,4 tháng ở nhóm giả dược so với 18,5 tháng ở nhóm Pertuzumab) (xem Bảng 1). Các kết quả nhất quán được ghi nhận ở các phân nhóm bệnh nhân đã được xác định trước bao gồm những phân nhóm dựa trên các yếu tố phân tầng vùng địa lý và đã điều trị hỗ trợ/tân hỗ trợ trước đó hoặc ung thư vú di căn mới.
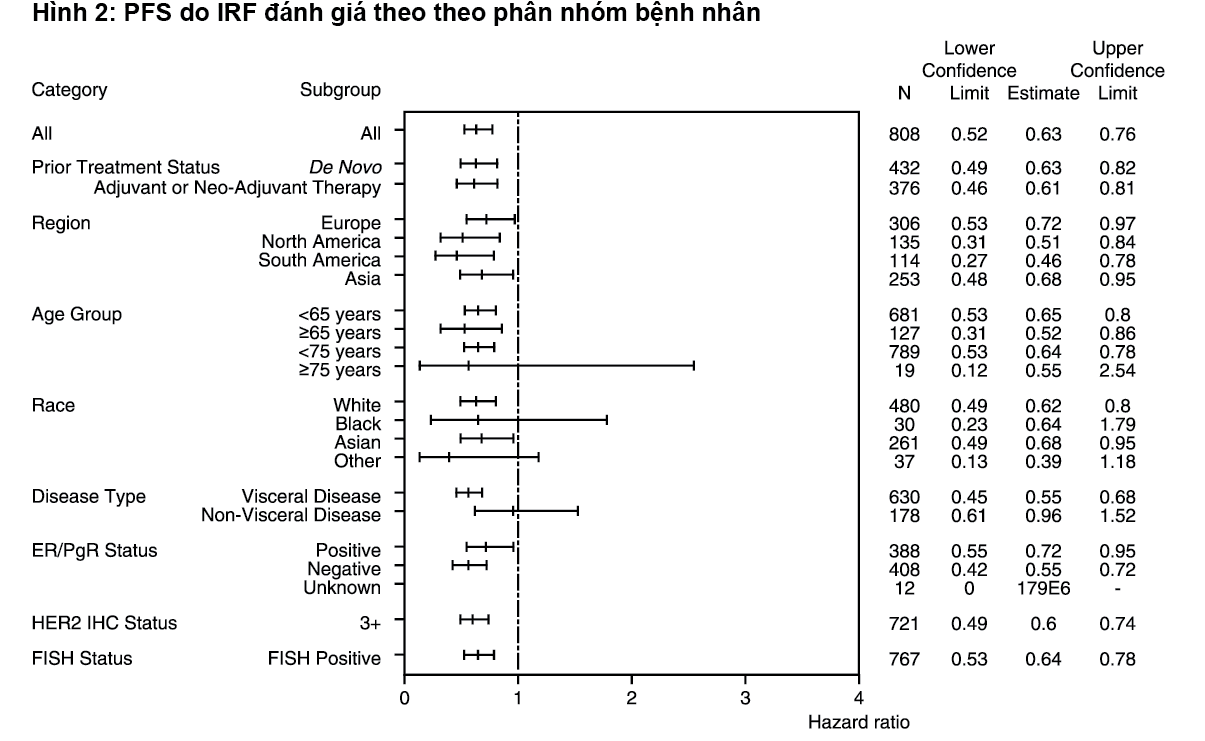
Các kết quả về hiệu quả điều trị từ nghiên cứu CLEOPATRA được tóm tắt trong Bảng 1.
– xem Bảng 1, Hình 1 và Hình 2.
| Bảng 1: Tóm tắt hiệu quả điều trị từ nghiên cứu CLEOPATRA | ||||
| Thông số | Giả dược + Herceptin + docetaxel n=406 | Pertuzumab + Herceptin + docetaxel n=402 | HR (95% Cl) | Giá trị p |
| Tiêu chí chính: | 0,62 | <0,0001 | ||
| Thời gian sống bệnh không tiến triển (do IRF đánh giá) | [0,51; 0,75] | |||
| Số lưạng bệnh nhãn bị một biến cố Trung vị tháng | 242 (59%) 12,4 | 191 (47,5%) 18,5 | ||
| Các tiêu chí phụ: | 221 (54,4%) 40,8 | 168(41,8%) | 0,68 | 0,0002 |
| Sống còn toàn bộ (kết quả phân tích cuối cùng OS) | 56,5 | [0,56; 0,84] | ||
| Số lưạng bệnh nhãn bị một biến cố* Trung vị tháng | ||||
| Tiêu chí phụ: | ||||
| Thời gian sống bệnh không tiến triển (đánh giá của nghiên cứu viên) | ||||
| Số lưạng bệnh nhãn bị một biến cố Trung vị tháng | 250 (61,6%) 12,4 | 201 (50,0%) 18,5 | 0,65 | <0,0001 |
| [0,54; 0,78] | ||||
| Tiêu chí phụ: | 336 | 343 | Khác biệt ORR: 10,8% | 0,0011 |
| Tỉ lệ đáp ứng khách quan (ORR) | 233 (69,3 %) [64,1; 74,2] | 275 (80,2 %) [75,6; 84,3] | [4,2; 17,5]% | |
| Số lưạng bệnh nhãn bị một biến cố Số bệnh nhãn đáp ứng’** | ||||
| 95% Cl cho ORR | ||||
| Đáp ứng hoàn toàn (CR) Đáp ứng một phần (PR) Bệnh ổn định (SD) | 14 (4,2 %) 219 (65,2 %) 70 (20,8 %) 28 (8,3 %) | 19(5,5%) 256 (74,6 %) 50 (14,6 %) 13(3,8%) | ||
| Bệnh tiến triển (PD) | ||||
| Thời gian đáp ứngA n = | 233 54,1 [46; 64] | 275 87,6 [71; 106] | ||
| Trung vị tuần 95% Cl cho Trung vị | ||||
* Phân tích cuối cùng về sống còn toàn bộ, ngày lấy dữ liệu phân tích 11/02/2014
** Các bệnh nhân có tỉ lệ đáp ứng toàn bộ tốt nhất của nhóm CR được khẳng định hoặc PR dựa trẽn RECIST.
^ Đánh giá ở những bệnh nhân có tỉ lệ đáp ứng toàn bộ tốt nhất của nhóm CR hoặc PR.
Tỉ lệ đáp ứng mục tiêu và thời gian đáp ứng dựa trên các phương pháp đánh giá khối u theo IRF.
Tại phân tích ban đầu về hiệu quả, kết quả phân tích gian kỳ của OS cho thấy chiều hướng lợi ích sống còn nghiêng mạnh về nhóm điều trị Perjeta.
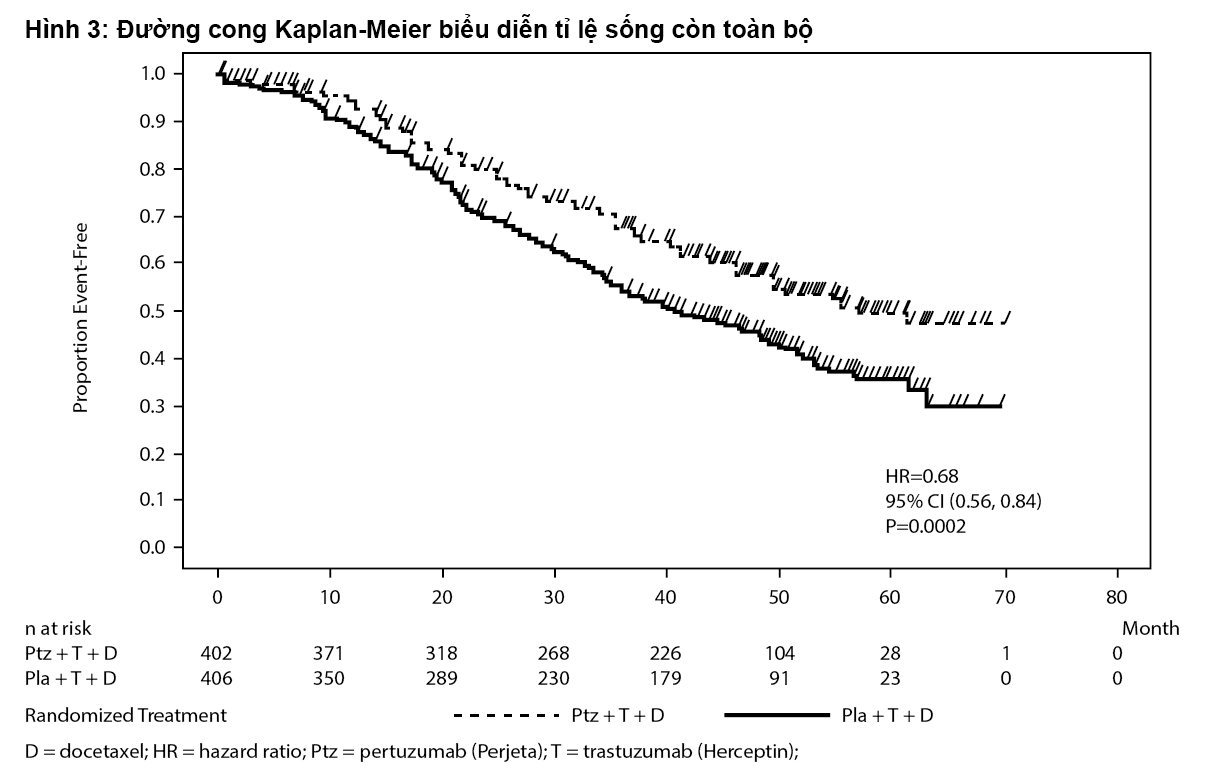
Kết quả phân tích gian kì OS, được thực hiện một năm sau phân tích sơ khởi về hiệu quả, đã chứng minh lợi ích về OS có ý nghĩa thống kê ở nhóm điều trị Perjeta (HR 0,66, p=0,0008 log-rank test). Trung vị thời gian đến khi tử vong ở nhóm dùng giả dược là 37,6 tháng trong khi vẫn chưa đạt được ở nhóm điều trị Perjeta.
Các phân tích cuối cùng OS được thực hiện khi 389 bệnh nhân đã chết (221 ở nhóm giả dược và 168 ở nhóm Perjeta). Lợi ích OS có ý nghĩa thống kê được duy trì ở nhóm Perjeta (HR 0,68, p=0,0002 test log-rank). Trung vị thời gian đến khi tử vong ở nhóm dùng giả dược là 40,8 tháng và 56,5 tháng ở nhóm dùng Perjeta (xem Bảng 1, Hình 3).
– xem Hình 3.
Không có sự khác biệt có ý nghĩa thống kê giữa hai nhóm điều trị về chất lượng cuộc sống liên quan đến sức khỏe đánh giá bằng thời gian đến khi triệu chứng tiến triển trên thang điểm phụ FACT-B TOI-PFB, định nghĩa là giảm 5 điểm trong thang điểm phụ (HR=0,97, 95% CI=0,81; 1,16). Trong một phân tích thăm dò, bệnh nhân điều trị với Pertuzumab phối hợp Herceptin và docetaxel có ít nguy cơ triệu chứng tiến triển theo thang điểm phụ ung thư vú FACT-B hơn (định nghĩa là giảm 2 điểm trong thang điểm phụ) so với những bệnh nhân điều trị Herceptin và docetaxel đơn lẻ (HR=0,78, 95% CI=0,65; 0,94).
BO17929
BO17929 là một nghiên cứu pha II, một nhóm quan sát, không phân nhóm ngẫu nhiên của Pertuzumab và được tiến hành trên những bệnh nhân HER2 (+) đã điều trị trước đó với Herceptin. Nghiên cứu được chia thành 3 nhóm thuần tập.
Nhóm 1 và 2: có 66 bệnh nhân trong nhóm 1 và 2 điều trị ít nhất một liều Pertuzumab và Herceptin (tất cả dân số điều trị và tất cả bệnh nhân đã điều trị di căn trước đó; một nửa bệnh nhân điều trị di căn theo phác đồ bước hai, trong khi 35% điều trị di căn theo phác đồ bước ba và hơn nữa. Ngoài ra, 71% đã dùng hóa trị liệu tân hỗ trợ). Tại thời điểm phân tích sơ khởi, trung vị thời gian điều trị trong nghiên cứu là 9 chu kỳ (27 tuần). Tại thời điểm phân tích sơ khởi, ORR và CBR được trình bày trong Bảng 2. Trung vị PFS và thời gian đến khi bệnh tiến triển (TTP) là 24 tuần. Trung vị thời gian đáp ứng là 11 tuần, và trung vị thời gian đáp ứng trong số những bệnh nhân đáp ứng là 25 tuần.
Nhóm 3: có 29 bệnh nhân điều trị ít nhất một chu kỳ với Pertuzumab. Trong số 29 bệnh nhân, có 12 người chỉ tham gia vào pha dùng đơn trị liệu, 17 người tiếp tục điều trị Pertuzumab và Herceptin khi có bằng chứng tiến triển bệnh khi dùng Pertuzumab đơn độc. Tất cả 29 bệnh nhân có bệnh tiến triển khi điều trị bước một cho bệnh di căn, và 41,4% cũng bị tiến triển sau khi điều trị bước hai. Tất cả bệnh nhân trong tập hợp 3 dùng ít nhất một liều đầy đủ Pertuzumab. Bệnh nhân dùng Pertuzumab và Herceptin điều trị trung bình tổng cộng 12 chu kỳ. Bảng 2 cho thấy việc điều trị Pertuzumab đơn lẻ có hoạt tính yếu ở những bệnh nhân đã thất bại với Herceptin (cột giữa). Các đáp ứng này xảy ra trên những bệnh nhân có tiến triển bệnh lý gần đây khi dùng riêng lẻ mỗi kháng thể. Ngoài ra có 3 bệnh nhân có bệnh ổn định kéo dài sáu tháng hoặc hơn với tỉ lệ có lợi ích lâm sàng nhìn chung là 35,3%.
| Bảng 2: Nghiên cứu B017929: dữ liệu về tính hiệu quả | |||
| Đáp ứng, n (%) | Nhóm 1 và 2 (Pertuzumab+Herceptin) (n=66) | Nhóm 3 (Pertuzumab đơn tri) (n=29) | Nhóm 3 (Pertuzumab+Hercepti n) (n=17) |
| Đáp ứng hoàn toàn (CR) | 4(6,1) | 0 (0,0) | 0 (0,0) |
| Đáp ứng một phần (PR) | 12(18,2) | 1 (3,4) | 3(17,6) |
| Tỉ lệ đáp ứng khách quan (ORR) | 16 (24,2) | 1 (3,4) | 3 (17,6) |
| Bệnh ổn định (SD) >6 tháng | 17(25,8) | 2 (6,9) | 3 (17,6) |
| Đáp ứng có lợi ích lâm sàng (CBR) | 33 (50,0) | 3(10,3) | 6 (35,3) |
| (CR + PR + SD >6 tháng) | |||
| Bệnh tiến triển (PD) | 33 (50,0) | 26 (89,7) | 9 (52,9) |
| Thiếu dữ liệu | 0 (0,0) | 0 (0,0) | 2 (11,8) |
| (không đánh giá đáp ứng) | |||
| Lưu ý: >6 tháng = 8 chu kỳ điều trị | |||
Điều trị tân bổ trợ ung thư vú
NEOSPHERE (WO20697)
NEOSPHERE là một nghiên cứu đa trung tâm, ngẫu nhiên pha II được tiến hành trên 417 bệnh nhân ung thư vú HER2 (+), tiến triển tại chỗ hoặc dạng viêm (T2-4d), có khả năng phẫu thuật được sắp lịch để tiến hành liệu pháp tân hỗ trợ. Mẫu khối u vú được tiến hành xét nghiêm sự biểu hiện quá mức HER2, được định nghĩa là IHC 3+ hay tỉ lệ khuếch đại ISH ≥ 2,0 được xác định bởi một phòng xét nghiệm trung tâm. Bệnh nhân được chọn ngẫu nhiên để điều trị một trong bốn phác đồ điều trị tân bổ trợ trước khi phẫu thuật như sau: Herceptin phối hợp với docetaxel, Pertuzumab phối hợp với Herceptin và docetaxel, Pertuzumab phối hợp với Herceptin, hoặc Pertuzumab phối hợp với docetaxel. Sự ngẫu nhiên được phân chia theo loại ung thư vú (có thể phẫu thuật được, tiến triển tại chỗ, hoặc dạng viêm) và tình trạng estrogen (ER) hoặc progesterone (PgR) dương tính.
Pertuzumab được tiêm truyền tĩnh mạch với liều ban đầu 840 mg, sau đó 420 mg mỗi ba tuần trong bốn chu kỳ. Herceptin được tiêm truyền tĩnh mạch với liều ban đầu là 8 mg/kg, tiếp theo là 6 mg/kg mỗi ba tuần trong bốn chu kỳ. Sau phẫu thuật, tất cả các bệnh nhân được điều trị ba chu kỳ 5-Fluorouracil (600 mg/m2), epirubicin (90 mg/m2), cyclophosphamid (600 mg/m2) (FEC) tiêm truyền tĩnh mạch mỗi ba tuần và Herceptin tiêm truyền tĩnh mạch mỗi ba tuần để hoàn thành một năm điều trị. Nhóm bệnh nhân dùng Pertuzumab phối hợp với Herceptin và docetaxel được tiêm truyền tĩnh mạch docetaxel mỗi ba tuần trong bốn chu kỳ trước khi dùng FEC sau khi phẫu thuật, do đó tất cả các bệnh nhân đều nhận được liều tích lũy tương đương với phác đồ hóa chất phối hợp với Herceptin.
Tiêu chí chính của nghiên cứu là tỷ lệ đáp ứng hoàn toàn về mô học (pCR) ở vú (ypT0/is). Tiêu chí phụ là tỷ lệ đáp ứng lâm sàng, tỷ lệ phẫu thuật bảo tồn vú (chỉ T2-3), thời gian sống không bệnh (DFS), và PFS. Tiêu chí thăm dò bổ sung là tỷ lệ pCR bao gồm tình trạng hạch (ypT0/isN0 và ypT0N0).
Về mặt đặc điểm bệnh nhân là cân bằng (tuổi trung bình là 49-50 tuổi, đa số là người da trắng (71%)) và tất cả đều là nữ. Tổng thể 7% bệnh nhân bị ung thư vú dạng viêm, 32% bệnh nhân ung thư vú tiến tiển tại chỗ và 61% ung thư vú có thể phẫu thuật được. Khoảng một nửa số bệnh nhân trong mỗi nhóm điều trị có tình trạng thụ thể hormon dương tính (được định nghĩa như ER dương tính và/hoặc PgR dương tính).
Kết quả nghiên cứu được trình bày trong Bảng 3. Có sự cải thiện có ý nghĩa lâm sàng và ý nghĩa thống kê tỉ lệ pCR (ypT0/is) đã được quan sát thấy ở những bệnh nhân dùng Pertuzumab phối hợp với Herceptin và docetaxel so với các bệnh nhân điều trị Herceptin phối hợp docetaxel (45,8% so với 29,0%, giá trị p=0,0141). Một hệ thống nhất quán của các kết quả đã được quan sát thấy bất kể định nghĩa pCR.
Tỷ lệ đáp ứng hoàn toàn về mô học (pCR) cũng như tầm quan trọng của cải thiện với Pertuzumab ở phân nhóm bệnh nhân có thụ thể hormon dương tính thấp hơn so với phân nhóm bệnh nhân có thụ thể hormon âm tính (5,9% với 26,0% và 27,3% với 63,2%, theo thứ tự).
TRYPHAENA (BO22280)
TRYPHAENA là một nghiên cứu lâm sàng đa trung tâm, ngẫu nhiên, Pha ll tiến hành trên 225 bệnh nhân ung thư vú HER2 dương tính, tiến triển tại chỗ, hoặc dạng viêm (T2-4d); có thể phẫu thuật. Mẫu khối u vú được xét nghiệm sự biểu hiện quá mức HER2, được định nghĩa là IHC 3+ hoặc khuếch đại ISH ≥ 2,0 được xác định bởi một phòng xét nghiệm trung tâm.
Bệnh nhân được chọn ngẫu nhiên để điều trị một trong ba phác đồ điều trị tân bổ trợ trước khi phẫu thuật như sau: 3 chu kỳ FEC, tiếp theo là 3 chu kỳ docetaxel, tất cả đều phối hợp với Pertuzumab và Herceptin; 3 chu kỳ của FEC đơn trị, tiếp theo là 3 chu kỳ docetaxel và Herceptin phối hợp với Pertuzumab, hoặc 6 chu kỳ TCH phối hợp với Pertuzumab. Sự ngẫu nhiên được phân chia theo loại ung thư vú (có thể phẫu thuật, tiến triển tại chỗ, hoặc viêm) và ER dương tính và/hoặc PgR dương tính.
Pertuzumab được tiêm truyền tĩnh mạch với liều khởi đầu là 840 mg, sau đó là 420 mg mỗi ba tuần. Herceptin được tiêm truyền tĩnh mạch với liều ban đầu là 8 mg/kg, sau đó là 6 mg/kg mỗi ba tuần. 5-Fluorouracil (500 mg/m2), epirubicin (100 mg/m2), cyclophosphamide (600 mg/m2) được tiêm truyền tĩnh mạch mỗi ba tuần trong 3 chu kỳ. Docetaxel được tiêm truyền tĩnh mạch với liều khởi đầu là 75 mg/m2 mỗi ba tuần với các lựa chọn tăng liều lên 100 mg/m2 tùy theo quyết định của bác sỹ điều trị nếu liều khởi đầu được dung nạp tốt. Tuy nhiên, ở nhóm Pertuzumab phối hợp với TCH, docetaxel được tiêm truyền tĩnh mạch 75 mg/m2 mà không tăng liều, và carboplatin (AUC 6) được tiêm truyền tĩnh mạch mỗi ba tuần. Sau phẫu thuật, tất cả các bệnh nhân được tiêm truyền tĩnh mạch Herceptin mỗi 3 tuần cho đủ một năm.
Tiêu chí chính của nghiên cứu là tính an toàn trên tim mạch trong suốt quá trình điều trị tân bổ trợ. Tiêu chí phụ là tỷ lệ pCR ở vú (ypT0/is), DFS, PFS và OS.
Về mặt đặc điểm bệnh nhân là cân bằng (tuổi trung bình là 49-50 tuổi, đa số là người da trắng (77%)) và tất cả đều là nữ. Tổng thể, 6% bệnh nhân bị ung thư vú dạng viêm, 25% là ung thư vú tiến triển tại chỗ và 69% là ung thư vú có thể phẫu thuật, với khoảng một nửa các bệnh nhân trong mỗi nhóm điều trị có tình trạng ER dương tính và/hoặc PgR dương tính.
Tỷ lệ pCR cao đã được quan sát thấy ở cả 3 nhóm điều trị (xem Bảng 3). Một hệ thống nhất quán của các kết quả đã được quan sát thấy bất kể định nghĩa pCR. Tỷ lệ đáp ứng hoàn toàn về mặt mô học (pCR) ở phân nhóm bệnh nhân có thụ thể hormon dương tính thấp hơn so với phân nhóm bệnh nhân có thụ thể hormon âm tính (46,2% với 50,0% và 65,0% với 83,8%, theo thứ tự).
| Bảng 3: Nghiên cứu NEOSPHERE (WO20697) và TRYPHAENA (BO22280): Tóm tắt hiệu quả (dân số dự định điều trị, ITT) | |||||||
| NEOSPHERE (WO20697) | TRYPHAENA (BO22280) | ||||||
| Thông số | T+D | Ptz+T+D | Ptz+T | Ptz+D | Ptz+T+FEC/ Ptz+T+D | FEC/ Ptz+T+D | Ptz+TCH |
| N=107 | N=107 | N=107 | N=96 | N=73 | N=75 | N=77 | |
| ypTO/is n(%) | 31 (29,0%) | 49 (45,8%) | 18(16,8%) | 23 (24,0%) | 45 (61,6%) | 43 (57,3%) | 51 (66,2%) |
| [95% khoảng tin cậy Cl]1 | [20,6; 38,5] | [36,1; 55,7] | [10,3; 25,3] | [15,8; 33,7] | [49,5; 72,8] | [45,4; 68,7] | [54,6; 76,6] |
| Khác biệt trong tỷ lệ pCR2 [95% khoảng tin cậy Cl]3 | +16,8% [3,5; 30,1] | -12,2% [-23,8; -0,5] | -21,8% [-35,1; -8,5] | Không có | Không có | Không có | |
| Giá trị p (với Simes corr. cho phép kiểm CMH)4 | 0,0141 (so với T+D) | 0,0198 (so với T+D) | 0,003 (so với Ptz+T+D) | Không có | Không có | Không có | |
| ypTO/is NO n(%) | 23 (21,5%) | 42 (39,3%) | 12(11,2%) | 17(17,7%) | 41 (56,2%) | 41 (54,7%) | 49 (63,6%) |
| [95% khoảng tin cậy Cl] | [14,1; 30,5] | [30,3; 49,2] | [5,9; 18,8] | [10,7; 26,8] | [44,1; 67,8] | [42,7; 66,2] | [51,9; 74,3] |
| ypTO NO n(%) | 13(12,1%) | 35 (32,7%) | 6 (5,6%) | 13(13,2%) | 37 (50,7%) | 34 (45,3%) | 40 (51,9%) |
| [95% khoảng tin cậy Cl] | [6,6; 19,9] | [24,0; 42,5] | [2,1; 11,8] | [7,4; 22,0] | [38,7; 62,6] | [33,8; 57,3] | [40,3; 63,5] |
| Đáp ứng lâm sàng5 | 79 (79,8%) | 89 (88,1%) | 69 (67,6%) | 65 (71,4%) | 67 (91,8%) | 71 (94,7%) | 69 (89,6%) |
T: Herceptin; D: docetaxel; Ptz: Pertuzumab; FEC: 5-fluorauracil, epirubicin, cyclophosphamide; TCI-
1.95% khoảng tin cậy Cl cho một mẫu nhị thức sử dụng phương pháp Pearson-Clopper.
2.Điêu trị Ptz + T + D và Ptz + T được so sánh với với T + D trong khi Ptz + D được so sánh với với Ptz + T + D
3.Khoảng 95% khoảng tin cậy Cl cho sự khác biệt của hal tỷ lệ sử dụng phương pháp Hauck-Anderson.
4.Glá trị p từ phép kiểm Cochran-Mantel-Haenszel, VỚI điều chỉnh bội số Slmes
5.Đáp ứng lâm sàng đạl diện cho những bệnh nhân có đáp ứng tổng thể tét nhất của CR hoặc PR trong quá trình điều trị tân bổ trợ (trong tổn thương vú ban đầu)
Cơ chế tác dụng:
Pertuzumab là một kháng thể đơn dòng tái tổ hợp nhân hóa gắn chuyên biệt vào vùng ngoại bào tham gia nhị trùng hóa (phân vùng II) của protein thụ thể yếu tố tăng trưởng thượng bì người (HER2), từ đó ngăn chặn sự bắt cặp dị hợp tử phụ thuộc ligand của HER2 với các HER khác cùng họ, bao gồm EGFR, HER3 và HER4. Kết quả là Pertuzumab ức chế sự phát tín hiệu nội bào của ligand thông qua hai con đường tín hiệu chính, men mitogen activated protein (MAP) kinase và phosphoinositide 3 kinase (PI3K). Việc ức chế hai con đường tín hiệu này lần lượt làm ngừng sự tăng trưởng tế bào và gây chết tế bào theo chương trình. Ngoài ra, Pertuzumab còn kích thích sự gây độc tế bào phụ thuộc kháng thể (ADCC).
Trong khi sử dụng Pertuzumab đơn lẻ gây ức chế sự tăng sinh tế bào khối u người, thì việc kết hợp Pertuzumab và Herceptin làm gia tăng đáng kể hoạt tính kháng khối u trong các mô hình ghép ngoại lai biểu hiện quá mức HER2.
[XEM TẠI ĐÂY]
5.2. Dược động học:
Xuyên suốt nhiều thử nghiệm lâm sàng với các chỉ định khác nhau, pertuzumab cho thấy không có thay đổi trong độ thanh thải thuốc ở liều 2-25 mg/kg. Dựa trên phân tính dược động học quần thể trên 481 bệnh nhân, độ thanh thải trung bình (CL) của pertuzumab là 0,235 l/ngày và thời gian bán thải trung bình là 18 ngày.
Phân tích dược động học quần thể cũng cho thấy không có sự khác biệt về tuổi, giới và chủng tộc (người Nhật Bản so với người không phải Nhật Bản). Kết quả albumin ban đầu và trọng lượng gầy của cơ thể là những đồng biến số quan trọng nhất ảnh hưởng tới CL. Độ thanh thải giảm ở những bệnh nhân có nồng độ albumin ban đầu cao hơn, và tăng ở những bệnh nhân có trọng lượng gầy cơ thể cao hơn. Tuy nhiên các phân tích độ nhạy thực hiện ở liều khuyến cáo và lịch trình điều trị của Pertuzumab cho thấy ở những giá trị cực đại của hai đồng biến số này, không có ảnh hưởng đáng kể nào lên khả năng đạt các nồng độ ở trạng thái ổn định, được xác định qua các mô hình tiền lâm sàng ghép khối u ngoại lai. Do đó, không cần điều chỉnh liều Pertuzumab dựa theo các đồng biến số này.
Kết quả dược động học của pertuzumab trong nghiên cứu NEOSPHERE đồng nhất với các dự đoán từ mô hình dược động học dân số trước đó.
Hấp thu: Pertuzumab được sử dụng qua đường truyền tĩnh mạch. Không thực hiện nghiên cứu với các đường dùng khác.
Phân phối: Xuyên suốt qua tất cả thử nghiệm lâm sàng, thể tích phân phối ở khoang trung tâm (Vc) và ngoại vi (Vp) trên bệnh nhân tiêu biểu lần lượt là 3,11 L và 2,46 L.
Chuyển hóa: Chuyển hóa của pertuzumab không được nghiên cứu trực tiếp. Các kháng thể được thải trừ chủ yếu bằng dị hóa.
Thải trừ: Trung vị độ thanh thải (CL) của pertuzumab là 0,235 L/ngày và trung vị thời gian bán thải là 18 ngày.
Dược động học trong các quần thể/dân số đặc biệt
Người lớn tuổi: Không có nghiên cứu chuyên biệt về pertuzumab trên bệnh nhân lớn tuổi. Trong một phân tích dược động học quần thể, tuổi tác không gây ảnh hưởng đáng kể đến dược động học của pertuzumab. Trong phân tích này, có 32,5% bệnh nhân (N=143) trên ≥ 65 tuổi và 9,1% bệnh nhân (N=40) trên ≥ 75 tuổi.
Suy thận: Không thực hiện nghiên cứu dược động học chính thức nào trên bệnh nhân suy thận. Dựa trên kết quả phân tích dược động học quần thể, suy thận dự kiến không gây ảnh hưởng tới sự phơi nhiễm với pertuzumab; tuy nhiên phân tích dược động học quần thể này chỉ bao gồm một số dữ liệu hạn chế về các bệnh nhân suy thận trung bình và nặng.
5.3 Giải thích:
Chưa có thông tin. Đang cập nhật.
5.4 Thay thế thuốc :
Chưa có thông tin. Đang cập nhật.
*Lưu ý:
Các thông tin về thuốc trên Pharmog.com chỉ mang tính chất tham khảo – Khi dùng thuốc cần tuyệt đối tuân theo theo hướng dẫn của Bác sĩ
Chúng tôi không chịu trách nhiệm về bất cứ hậu quả nào xảy ra do tự ý dùng thuốc dựa theo các thông tin trên Pharmog.com
6. Phần thông tin kèm theo của thuốc:
6.1. Danh mục tá dược:
Glacial acetic acid , L-Histidine, Sucrose, Polysorbate 20, Water for injections
6.2. Tương kỵ :
Không thấy có tương kỵ nào giữa Pertuzumab và polyvinylchloride, polyethylene hoặc túi polyolefin không PVC.
Không nên dùng dung dịch dextrose (5%) để pha loãng Pertuzumab bởi vì tính chất hóa lý của thuốc không ổn định trong những dung dịch này (dung dịch pertuzumab pha loãng trong các túi D5W truyền tĩnh mạch không duy trì được pH ổn định sau khi bảo quản ở nhiệt độ phòng (27-33°C) 24 giờ tiếp theo là bảo quản ở nhiệt độ tủ lạnh trong 24 giờ [2-8°C]). Không nên trộn hoặc pha Pertuzumab với các thuốc khác.
6.3. Bảo quản:
Bảo quản lọ thuốc trong tủ lạnh ở nhiệt độ 2°C-8°C.
Giữ lọ thuốc trong vỏ hộp để tránh ánh sáng.
Không để đông lạnh. không lắc lọ thuốc.
Hạn dùng của dung dịch truyền chứa thuốc
Thuốc Pertuzumab không chứa chất bảo quản kháng khuẩn; do đó cần thận trọng để đảm bảo tính vô khuẩn của dung dịch đã pha.
Dung dịch truyền Pertuzumab được pha loãng trong túi polyvinylchloride (PVC) hoặc polyolefin không PVC chứa dung dịch truyền NaCl 0.9% theo tiêu chuẩn Dược điển Mỹ (USP), có thể bảo quản đến 24 giờ ở nhiệt độ 2°C-8°C (36°F-46°F) trước khi sử dụng. Pertuzumab được pha loãng ổn định trong 24 giờ (ở nhiệt độ tới 30°C). Tuy nhiên bởi vì Pertuzumab pha loãng không có chất bảo quản, nên dung dịch pha loãng nên được bảo quản lạnh (2°C-8°C).
6.4. Thông tin khác :
Tính sinh miễn dịch
Các bệnh nhân trong thử nghiệm then chốt CLEOPATRA được kiểm tra tại nhiều thời điểm để tìm các kháng thể kháng điều trị (ATA) với Pertuzumab. Có khoảng 6,2% bệnh nhân (23/372) trong nhóm giả dược và 2,8% bệnh nhân (11/386) điều trị Pertuzumab được kiểm tra có kết quả dương tính với ATA. Không có bệnh nhân nào trong số 34 bệnh nhân này xảy ra các phản ứng quá mẫn/sốc phản vệ liên quan rõ rệt tới ATA.
Các kết quả định lượng tính sinh miễn dịch phụ thuộc rất nhiều vào một số yếu tố như độ nhạy và tính đặc hiệu, phương pháp, xử lý mẫu, thời gian thu thập mẫu, các thuốc dùng đồng thời và bệnh chính. Vì những lý do này nên việc so sánh tỉ lệ kháng thể kháng Pertuzumab với tỉ lệ kháng thể kháng các thuốc khác có thể không chính xác.
Tính an toàn tiền lâm sàng
Tính gây ung thư: Không thực hiện các nghiên cứu dài hạn trên động vật để đánh giá độc tính gây ung thư của pertuzumab.
Tính sinh đột biến: Không thực hiện các nghiên cứu đánh giá độc tính gây đột biến của pertuzumab.
Suy giảm khả năng sinh sản: Không thực hiện nghiên cứu chuyên biệt trên hệ sinh sản ở các động vật để đánh giá tác động của Pertuzumab. Không quan sát thấy tác dụng ngoại ý trên cơ quan sinh sản của khỉ cynomolgus đực và cái trong các nghiên cứu độc tính với liều lặp lại kéo dài tới sáu tháng.
Tính gây quái thai: Các nghiên cứu độc tính sinh sản đã được thực hiện trên khỉ cynomolgus với liều ban đầu từ 30 tới 150 mg/kg và liều duy trì từ 10 tới 100 mg/kg để đạt được các mức phơi nhiễm tương ứng với trên lâm sàng. Pertuzumab truyền tĩnh mạch từ ngày mang thai thứ 19 cho tới ngày thứ 50 (giai đoạn hình thành các cơ quan) cho thấy có độc tính phôi phụ thuộc liều, trong đó liều tăng làm chết phôi thai/thai trong thời gian giữa ngày thai kỳ thứ 25 tới 70. Làm chậm phát triển thận và thiểu ối được phát hiện vào ngày thai kỳ thứ 100.
Khác: Trên khỉ cynomolgus, truyền tĩnh mạch pertuzumab mỗi tuần với liều lên tới 150 mg/kg/liều được dung nạp tốt. Với những liều 15 mg/kg và cao hơn, ghi nhận có xảy ra tiêu chảy liên quan tới điều trị với mức độ nhẹ và không thường xuyên. Trong nhóm phụ những con khỉ dùng liều kéo dài (liều mỗi 7 tới 26 tuần) xảy ra các đợt tiêu chảy mất nước, được xử trí bằng bù nước qua truyền tĩnh mạch.
6.5 Tài liệu tham khảo:
Dược Thư Quốc Gia Việt Nam
HDSD Thuốc.
7. Người đăng tải /Tác giả:
Bài viết được sưu tầm hoặc viết bởi: Bác sĩ nhi khoa – Đỗ Mỹ Linh.
Kiểm duyệt , hiệu đính và đăng tải: PHARMOG TEAM